1710
Validation of Diffusion MRI Biomarkers of Peripheral Nerve Degeneration in ALS1Translational Neuroscience, Barrow Neurological Institute, Phoenix, AZ, United States, 2Biomedical Sciences, Creighton University, Omaha, NE, United States, 3School of Biological and Health System Engineering, Arizona State University, Phoenix, AZ, United States, 4Cancer Systems Imaging, University of Texas MD Anderson Cancer Center, Houston, TX, United States
Synopsis
Keywords: Nerves, Diffusion Tensor Imaging
ALS is a fatal, neurodegenerative disease that affects motor neurons. Assessment of disease progression is difficult and relies on tests that lack specificity/responsiveness. While imaging biomarkers may meet this need, previous work has focused on the motor neurons or skeletal muscles, with nerves that form the link between these tissues being largely understudied. Previous studies suggest DTI in human nerves may be sensitive to nerve degeneration in ALS and here we sought to validate these measures in a rat model. Preliminary findings showed a progressive decrease in FA, which correlated to the onset of motor neuron loss and muscle wasting.Animal Model
Rats were chosen because their sciatic nerves are large enough for imaging and have a similar structure and morphology to humans. SOD1-G93A rats were chosen as they are an ALS model with known disease progression. [5] Timepoints were chosen over a range from before disease progression becomes clinically detectable to advanced disease stage (110, 150, and 170 days post-birth). In this pilot study, the sciatic nerve from at least 3 animals per cohort (control and SOD1) at each time-point were extracted for ex vivo DTI.
Sample preparation
Post-euthanasia, nerves were removed and immersed in 3% glutaraldehyde/2% paraformaldehyde for 24 hours. Following one week of washing in phosphate buffered saline (PBS), nerves were immersed in 1 mM Gd-DTPA (Magnevist; Berlex, Montville, NJ) at 4 °C for a minimum of 36 hours to reduce spin-lattice relaxation times. Nerves were then trimmed to approximately 1 cm in length and excess water removed before samples were placed in 3mm capillary tubes filled with perfluorcarbon solution (Fomblin; Solvay, Thorofare, NJ) to prevent sample dehydration without affecting MR signal.
MRI protocol
Groups of three nerves were arranged in an asymmetric pattern in a custom 3D printed holder and scanned simultaneously. Diffusion-weighted MRI data were acquired in a 7-T, 70/30 Bruker Biospec with AVANCE III electronics using Paravision 5.1 (Rheinstetten, Germany) and a 30-mm mouse volume quadrature coil (Bruker Rheinstetten, Germany) for transmission and reception. Images were acquired with a three-dimensional diffusion-weighted spin-echo sequence and the following parameters: field-of-view = 1mm × 1mm × 1 mm3, resolution= 125 × 125 × 370 μm3, TE/TR = 22/425 ms, gradient pulse duration/diffusion time (δ/Δ) = 4/12 ms, b-value = 2000 s/mm2, 20 diffusion directions, number of averaged excitations = 2, and scan time = 11 hours and 43 minutes. MRI analysis. Diffusion tensors were estimated on voxel-wise basis using a weighted linear least-squares estimation in MATLAB. The following indices were estimated from the diffusion tensor: FA, MD, RD, AD. Regions-of-interest (ROI) were drawn manually on at least 2 consecutive slices in each nerve to calculate the mean diffusion parameters. Statistical analysis. Statistical analyses were performed in MATLAB. An unpaired student’s t-test was used to compare pairwise differences between the cohorts (control/SOD1) at each timepoint for each DTI parameter. All p-values were adjusted for the effect of multiple comparisons. [6]
Results/Discussion
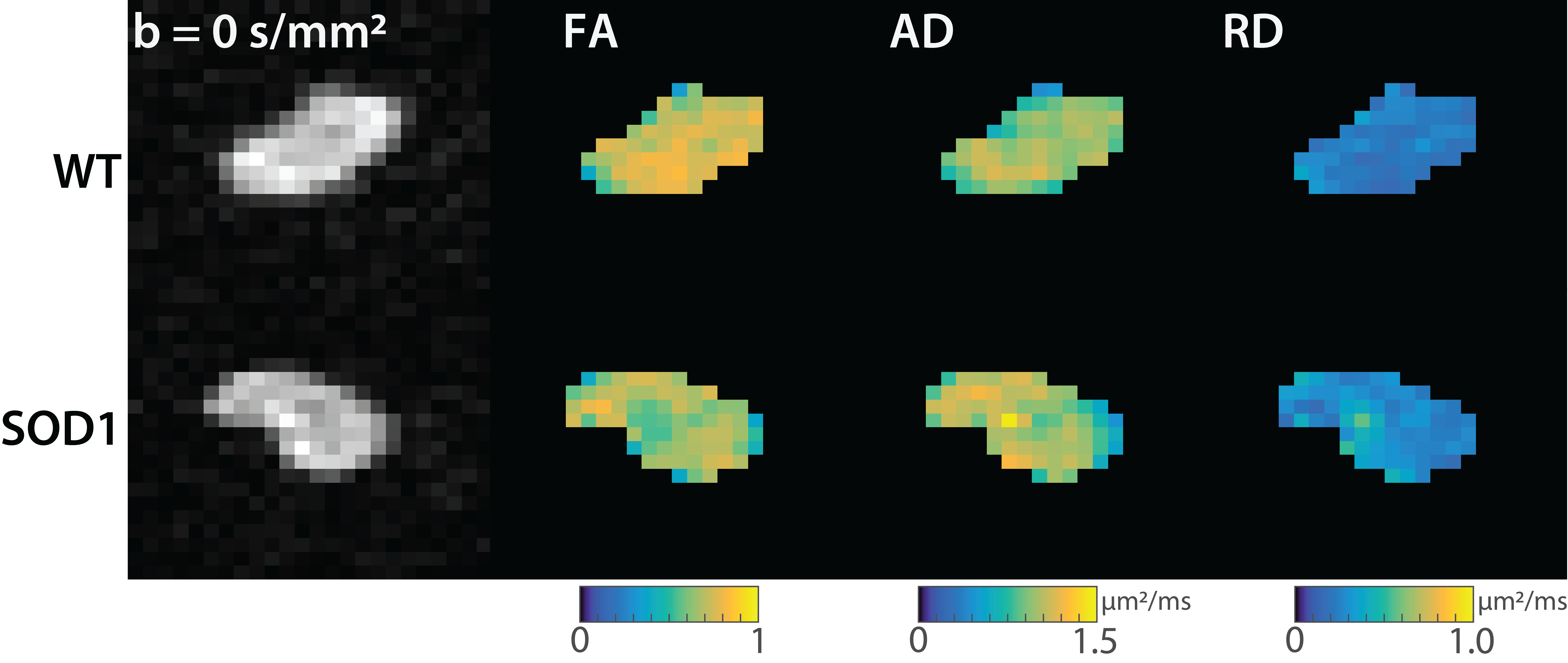
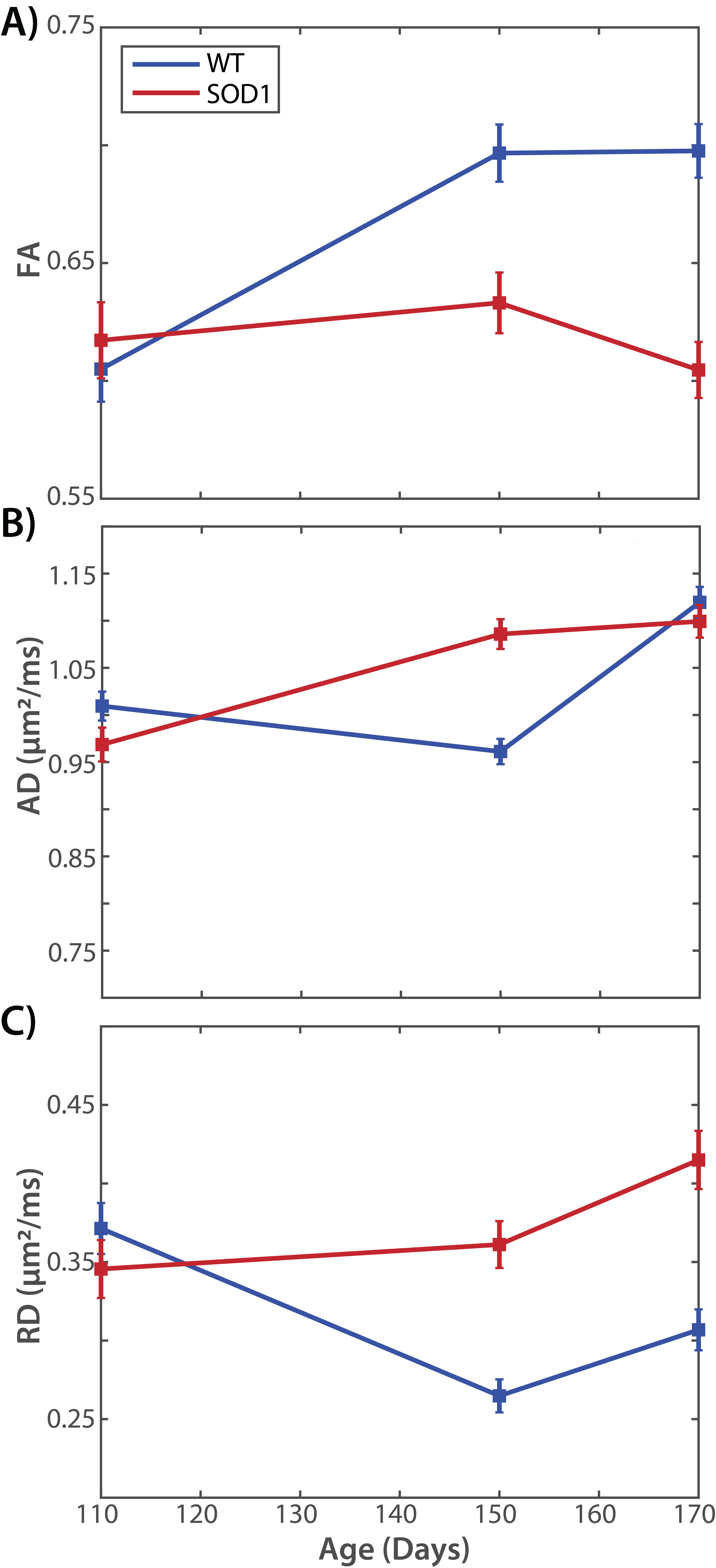
Figure 1 shows representative images and parameter maps from the sciatic nerve of the wild type (top) and SOD1 (bottom) rats at 170 days. Note the reduced FA values in the SOD1 nerves at this time relative to the wild type nerves, which was primarily driven by elevated RD values in the SOD1 nerves. Figure 2 summarizes the evolution of the mean diffusion parameters (FA, AD, RD) across time. At 110 days, no difference was observed between the two cohorts. However, by 150 days, a significant reduction in FA values was observed in SOD1 rats, which was more pronounced by 170 days. Overall, this was driven primarily by changes in RD, which is consistent with our previous studies in rat models of nerve trauma [7], which indicated FA changes in degenerating nerves are primarily related to changes in axonal density. These findings are also consistent with previous human studies, [4] which found a FA values decreased over time and related to changes in electrophysiological measures of axonal loss (MUNE) in humans with ALS.
Together, these findings suggest that DTI-derived FA values may be pathologically valid biomarkers of nerve degeneration in ALS. Future work will focus on additional histopathological studies aimed at providing this validation and the investigation of advanced diffusion models that may provide higher levels of pathological specificity relative to DTI (at the expense of longer scan times).
Acknowledgements
No acknowledgement found.References
1. Westeneng, H.J., et al., Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. Lancet Neurol, 2018. 17(5): p. 423-433.
2. Chiò, A., et al., Global Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review of the Published Literature. Neuroepidemiology, 2013. 41(2): p. 118-130.
3. Wong, C., et al., Clinical trials in amyotrophic lateral sclerosis: a systematic review and perspective. Brain Communications, 2021. 3(4).
4. Simon, N.G., et al., Peripheral nerve diffusion tensor imaging as a measure of disease progression in ALS. Journal of Neurology, 2017. 264(5): p. 882-890.
5. Matsumoto, A., et al., Disease progression of human SOD1 (G93A) transgenic ALS model rats. J Neurosci Res, 2006. 83(1): p. 119-33.
6. Benjamini, Y. and Y. Hochberg, Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological), 1995. 57(1): p. 289-300.
7. Manzanera Esteve, I.V., et al., Probabilistic Assessment of Nerve Regeneration with Diffusion MRI in Rat Models of Peripheral Nerve Trauma. Scientific Reports, 2019. 9(1).
Figures