2322
In vivo imaging of bone marrow endothelial dysfunction promoting myeloid cell expansion in cardiovascular disease1Center for Systems Biology, Massachusetts General Hospital and Harvard Medical School, Boston, MA, United States, 2Biomedical Engineering, Technion, Israel Institute of Technology, Haifa, Israel, 3Max Planck Institute for Molecular Biomedicine, Muenster, Germany, 4Institut del Cor Germans Trias i Pujol, Barcelona, Spain, 5Genetics, Harvard Medical School, Boston, MA, United States, 6State Key Laboratory of Cell Biology, CAS Center for Excellence in Molecular Cell Science, Institute of Biochemistry and Cell Biology, Chinese Academic of Sciences, Shanghai, China, 7Cardiovascular Research Institute, Department of Molecular Physiology and Biophysics, Baylor College of Medicine, Houston, TX, United States, 8Institute of Neurosciences and Department of Cellular Biology, Physiology and Immunology, Universitat Autònoma de Barcelona, Barcelona, Spain, 9Institute for Molecular Science of Medicine, Aichi Medical University, Aichi, Japan, 10Wellman Center for Photomedicine, Massachusetts General Hospital and Harvard Medical School, Boston, MA, United States, 11Heart Center, Department of Cardiology, Amsterdam University Medical Center, University of Amsterdam, Amsterdam, Amsterdam, Netherlands, 12Center for Regenerative Medicine and Cancer Center, Massachusetts General Hospital, Boston, MA, United States, 13Stem Cell and Regenerative Biology, Harvard University, Cambridge, MA, United States, 14Martinos Center for Biomedical Imaging, Department of Radiolog, Massachusetts General Hospital and Harvard Medical School, Charlestown, MA, United States, 15Pittsburgh Heart, Lung, Blood and Vascular Medicine Institute, Division of Cardiology, Department of Medicine, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States, 16Pathology, Massachusetts General Hospital and Harvard Medical School, Boston, MA, United States, 17Division of Cardiovascular Medicine, Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, United States, 18Cardiology, Angiology and Pneumology, Heidelberg University Hospital, Heidelberg, Germany
Synopsis
Hematopoietic stem cells at the bone marrow (BM) niche generates excess inflammatory leukocytes harming the heart after a myocardial infarction (MI). Whether MI affects the hematopoietic organ’s microvasculature is unknown. Here intravital microscopy shows that MI triggers endothelial dysfunction, leakage, and angiogenesis in the BM, leading to systemic leukocytosis. These novel findings were imaged by noninvasive PET imaging of integrin αVβ3 activation concomitant with measuring leakiness using dynamic contrast enhanced MRI. Endothelial deletion of Vegf receptor 2 (Vegfr2) curbed emergency hematopoiesis after MI. Our findings establish that MI remodels the vascular BM niche, stimulating hematopoiesis and production of inflammatory leukocytes.
Introduction
Increased leukocytosis, associates with worse cardiovascular outcomes in large clinical cohorts1. Specifically, bone marrow (BM) derived monocytes and neutrophils accumulate in the failing myocardium. Once recruited, leukocytes release inflammatory cytokines and proteases that destabilize inducing heart failure2. Because myeloid cells, like monocytes and neutrophils, circulate only for a few days after release from the BM3, examining the pathways that instigate their overproduction in cardiovascular disease (CVD) may provide a key to curbing cardiovascular morbidity. Central to the pathogenesis of ischemia, are structural and functional vascular changes which occur system-wide. Yet, how this condition affects the BM vasculature, which regulates hematopoietic stem and progenitor (HSPC) activity and leukocyte trafficking4, is unknown.Methods
We studied the BM vasculature in mice with acute MI. We examined the femoral vasculature with immunofluorescence and flow cytometry. Next, we performed intravital microscopy at the skull before and after MI to examine structural and functional changes, specifically (i) angiogenesis, (ii) integrin activation, (iii) vascular leakage and (iv) blood flow. (i) We imaged AplnCreER;Rosa26ZsGreen/+ reporter mice5, in which sprouting endothelial cells and their progeny express ZsGreen. We visualized (ii) fluorescent RGD probe that binds to integrin αVβ3. We studied (iii) endothelial barrier function with intravital time lapse imaging after labeling the blood pool with fluorescent albumin6. We assayed (iv) blood velocity in the skull BM. Next, we translated our microscopy studies towards non-invasive whole mouse imaging, enabling system-wide in vivo assessment of all hematopoietic sites. Therefore, an RGD-containing gallium-68 PET reporter (68Ga-RGD) and a gadolinium-labeled albumin derivative were co-injected into mice with MI, followed by a joined PET and dynamic contrast enhanced MRI protocol6. PET data were validated with autoradiography and scintillation counting. Finally, we used two mouse models: (i) anti-Vegfr2 antibody treatment in mice after MI; (ii) Cdh5CreERT2;Kdrfl/fl animals, in which endothelial cells lack expression of Vegrf2. We performed immunofluorescence, flow cytometry and intravital microscopy for permeability and integrin activation in these two models. Data are displayed as mean±SEM. Statistical analysis was done using two-tailed Mann-Whitney unless otherwise stated.Results
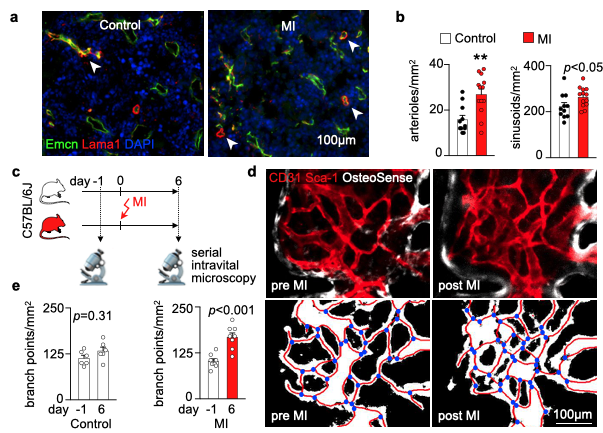
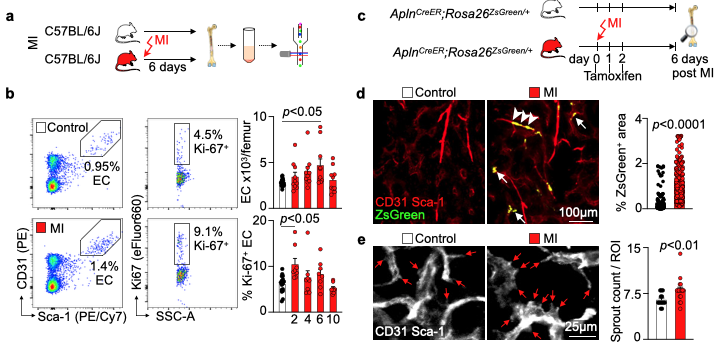
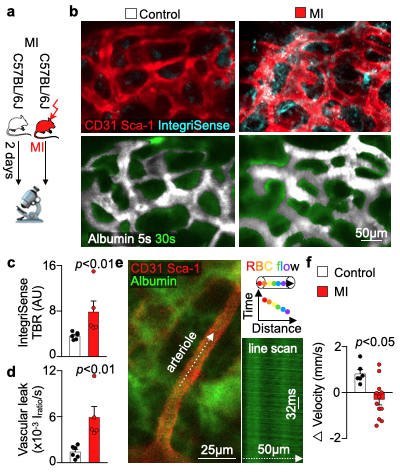
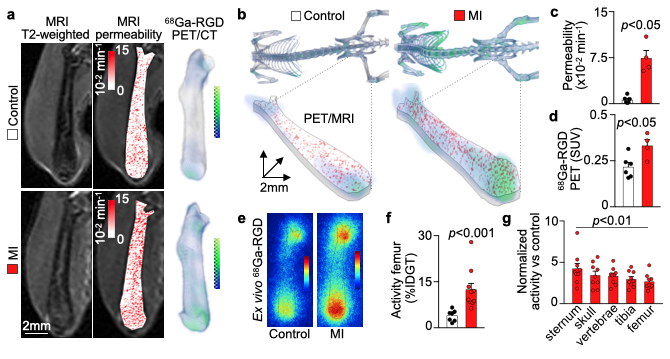
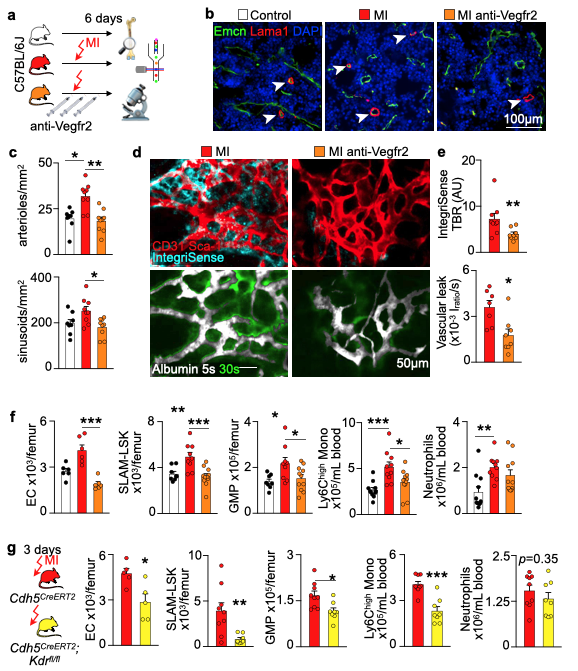
We hypothesized that post-MI angiogenesis in the bone marrow facilitates emergency hematopoiesis, perhaps by expanding the vascular niche in which myelopoiesis occurs. We detected in mice after MI increased BM vessel density (Fig. 1a,b). Serial intravital microscopy documented increasing marrow vascularity post-MI (Fig. 1c-e). Flow cytometry revealed an increased number of BM endothelial cells (BMEC) and their proliferation rates with a peak at day 6 after MI(Fig. 2a, b)6. Using confocal imaging on day 6 post-MI, we identified newly developed ZsGreen+ BM vessels (Fig. 2d) together with an increased amount of vascular sprouts (Fig. 2e). We observed in mice at day 2 after MI integrin activation and accelerated vascular leakage (Fig. 3a-d). To probe endothelial dysfunction, we next quantified blood velocity in skull marrow microvessels after intravenous acetylcholine injection. For healthy vasculature7, acetylcholine increased velocity in BM arterioles of control mice and decreased velocity decreased in mice post-MI (Fig. 3 e, f).Next, we complemented our intravital microscopy studies with non-invasive whole mouse imaging, enabling system-wide in vivo assessment of all hematopoietic sites. Using 68Ga-RGD PET and a gadolinium-labeled albumin, which relied on imaging agents corresponding to the fluorescent companions used for intravital microscopy, indicated increased integrin binding and higher vascular permeability in the femur after MI (Fig. 4a-d). Autoradiography (Fig. 4e) and scintillation counting (Fig. 4f) confirmed higher 68Ga-RGD accumulation in the bones of mice with acute MI. Analysis of 68Ga-RGD in various hematopoietic sites identified the highest activity in the sternum (Fig. 4g), possibly due to its proximity to the infarcted heart.We next hypothesized that post-MI angiogenesis in the BM facilitates emergency hematopoiesis, perhaps by expanding the vascular niche in which myelopoiesis occurs. Inhibiting the cognate receptor Vegfr2 with a neutralizing antibody abolished the post-MI increase in BM arterioles, and sinusoids (Fig. 5a-c). In line with these ex vivo data, intravital microscopy detected reduced integrin presence and less vascular leakage, which increase during angiogenesis45, in post-MI mice treated with Vegfr2 blocking antibody (Fig. 5d, e). More importantly, Vegfr2 inhibition after MI reduced the amount of BMECs and promoted HSPC quiescence and lowered the amount of circulating Ly6Chigh monocytes (Fig. 5f). To verify if reduced myelopoiesis could be secondary by inhibiting BM angiogenesis or alternatively caused by blocking the Vegfr2 receptor on hematopoietic cells52, we bred Cdh5CreERT2 with Kdrfl/fl mice for tamoxifen-inducible, endothelial cell-specific deletion of Vegfr2. In these mice, in which baseline hematopoiesis was unaffected, post-MI hematopoiesis and circulating Ly6Chigh monocytes (Fig. 5g) declined, indicating that Vegf signaling affects leukocytosis via BM endothelial cells.Discussion
The changes in BMEC structure and function may facilitate trafficking of cells and signals across the BM endothelium, potentially supporting HSPC activation, processes that augment leukocytosis. Our data on BMEC Vegfr2 is supported by prior reports that Vegfr2 is essential for vascular recovery after irradiation8, as well as clinical data in patients after MI have high blood VEGF levels9.Conclusion
In vivo imaging revealed that cardiovascular disease affects the BM vasculature's structure and, most importantly, expands the vascular niche after MI and this effect is mediated by endothelial Vegfr2.Acknowledgements
The authors thank Robert S. Fujinami (University of Utah School of Medicine, Salt Lake City, UT, USA) for sharing mouse strains, the MGH Mouse Imaging Program for assistance with imaging, Hye-Yeong Kim for radiolabeling imaging tracers, the Center for Skeletal Research Core (NIH P30 AR066261) for histological processing, the HSCI-CRM Flow Cytometry Core for assistance with cell sorting and Kaley Joyes for editing the manuscript. This work was funded in part by U.S. federal funds from the National Institutes of Health (HL142494, HL139598, HL128264, HL131478, HL125428 and HL131495), the Global Research Lab (GRL) program (NRF-2015K1A1A2028228) within the National Research Foundation by the Korean government, the MGH Research Scholar Program; the Deutsche Forschungsgemeinschaft (RO5071/1-1 and HO 5279/1-2) and the Ministerio de Economía y Competitividad y Fondo Europeo de Desarrollo Regional (SAF2014-56546-R and RTI2018-101105-B-I00).References
1. Coller, B. S. Leukocytosis and ischemic vascular disease morbidity and mortality: Is it time to intervene? Arteriosclerosis, Thrombosis, and Vascular Biology vol. 25 658–670 (2005).
2. Swirski, F. K. & Nahrendorf, M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nature Reviews Immunology vol. 18 733–744 (2018).
3. Patel, A. A. et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med. 214, 1913–1923 (2017).
4. Itkin, T. et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature (2016) doi:10.1038/nature17624.
5. Liu, Q. et al. Genetic targeting of sprouting angiogenesis using Apln-CreER. Nat. Commun. 6, 1–12 (2015).
6. Vandoorne, K. et al. Imaging the vascular bone marrow niche during inflammatory stress. Circ. Res. 123, 415–427 (2018).
7. Flammer, A. J. et al. The Assessment of Endothelial Function – From Research into Clinical Practice. Circulation 126, 753 (2012).
8. Hooper, A. T. et al. Engraftment and Reconstitution of Hematopoiesis Is Dependent on VEGFR2-Mediated Regeneration of Sinusoidal Endothelial Cells. Cell Stem Cell 4, 263–274 (2009).
9. Kranz, A., Rau, C., Kochs, M. & Waltenberger, J. Elevation of vascular endothelial growth factor A serum levels following acute myocardial infarction. Evidence for its origin and functional significance. J. Mol. Cell. Cardiol. (2000) doi:10.1006/jmcc.1999.1062.
Figures