0056
Glycogen accumulation in the brain of classic-infantile Pompe patient measured with single-voxel 1H MRS and 2D-MRSI at 7T1C.J. Gorter Center for High Field MRI, Department of Radiology, Leiden, Netherlands, 2Danish Research Centre for Magnetic Resonance, Centre for Functional and Diagnostic Imaging and Research, Copenhagen University Hospital Amager and Hvidovre, Copenhagen, Denmark, 3Center for Lysosomal and Metabolic diseases, Department of Neurology, Erasmus MC University Medical Center, Rotterdam, Netherlands, 4Center for Lysosomal and Metabolic diseases, Department of Pediatrics, Erasmus MC University Medical Center, Rotterdam, Netherlands
Synopsis
Pompe disease is caused by an abnormal accumulation of glycogen in the lysosomes of multiple tissues including the brain due to a deficit in acid α-glucosidase (GAA). The development of enzyme replacement therapy with recombinant human GAA (rhGAA) has dramatically improved patients’ survival, however, rhGAA does not reach the brain which remains untreated. Consequently, classic-infantile Pompe patients may develop progressive white matter lesions and cognitive problems. Here, we used single-voxel 1H MRS and spectroscopic imaging and found an accumulation of glycogen and significant decrease in total-N-acetyl-aspartate in the brain of classic-infantile patients (n=3) when compared to age-matched healthy controls (n=3).
Introduction
Glycogen storage disease type II, also known as Pompe disease, is a rare autosomal recessive disorder caused by a deficit in the lysosomal glycogen degradation enzyme, acid α-glucosidase (GAA)1. Classic-infantile Pompe patients are the most affected and do not reach the age of one year, if untreated. Enzyme replacement therapy with recombinant human acid α-glucosidase (rhGAA) has dramatically improved patient survival2. rhGAA, however, does not cross the blood-brain-barrier (BBB) and thus the brain remains unaffected by the treatment. As classic-infantile patients get older, a new phenotype appears, showing hampering or even decline of cognitive development3,4. Conventional brain imaging has shown white matter (WM) lesions in the brain of classic-infantile patients, which progressively spread with increasing age3,4. Post-mortem studies in Pompe disease demonstrated that glycogen accumulates in lysosomes within neurons of the cerebral cortex, brainstem, and spinal cord and in glial cells of the WM and the cerebral cortex5. This suggests that cell-specific neurochemical profile might be affected in Pompe disease. Here, we investigated the potential of single-voxel 1H MRS (SVS) and spectroscopic imaging (MRSI) at 7T to provide a non-invasive readout of glycogen accumulation and other metabolic alterations in the brain of classic-infantile Pompe patients.Materials and methods
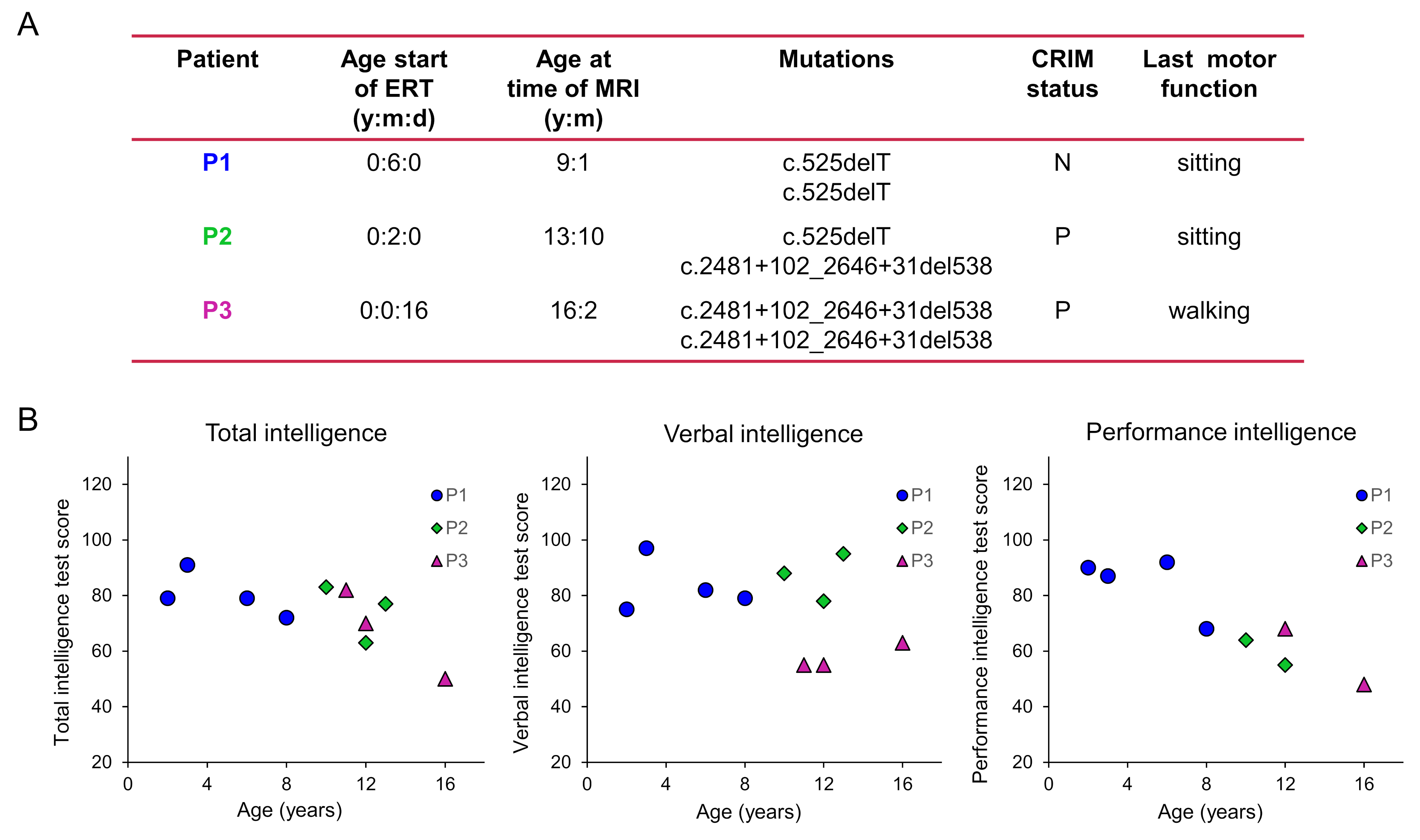
Subjects: Three classic-infantile patients (9, 14, and 16yo) and three age-matched healthy controls participated in this study so far. Patient diagnosis was confirmed by enzyme activity assays and mutation analysis in the first 6 months after birth, and patients were treated with a dose of rhGAA of 40mg/kg/week at the time of the MRI/MRS scans. Patient characteristics and longitudinal neuropsychological evaluations are given in Fig.1. MRI performed at 3T showed that all patients already presented abnormal WM lesions one year before our study. Study protocols were approved by the institutional review board and written informed consent was obtained.MR setup: Experiments were conducted on a 7T whole-body MRI scanner (Philips Healthcare, The Netherlands) equipped with a volume transmit/32-channel receive head coil (Nova Medical, USA).
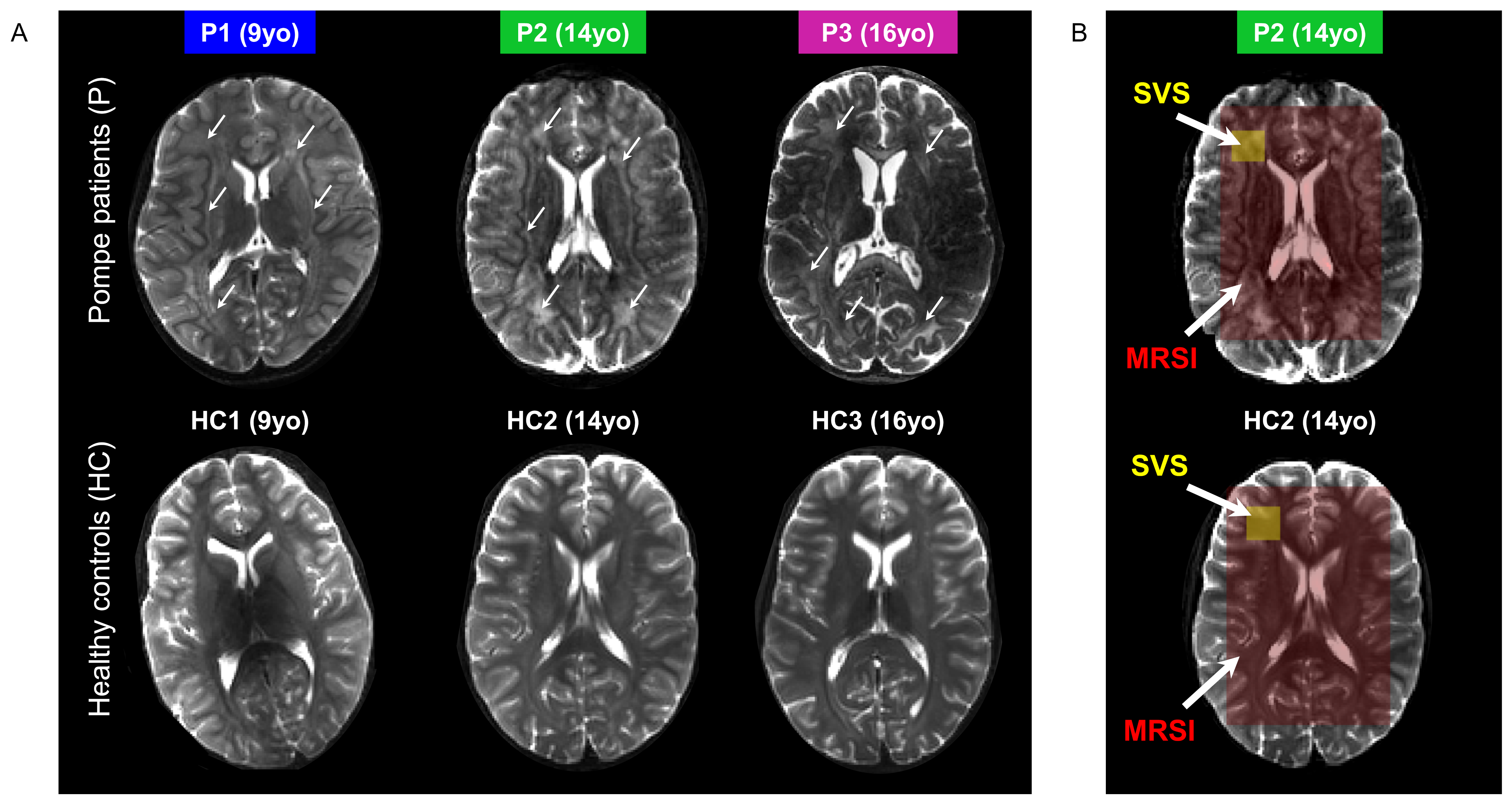
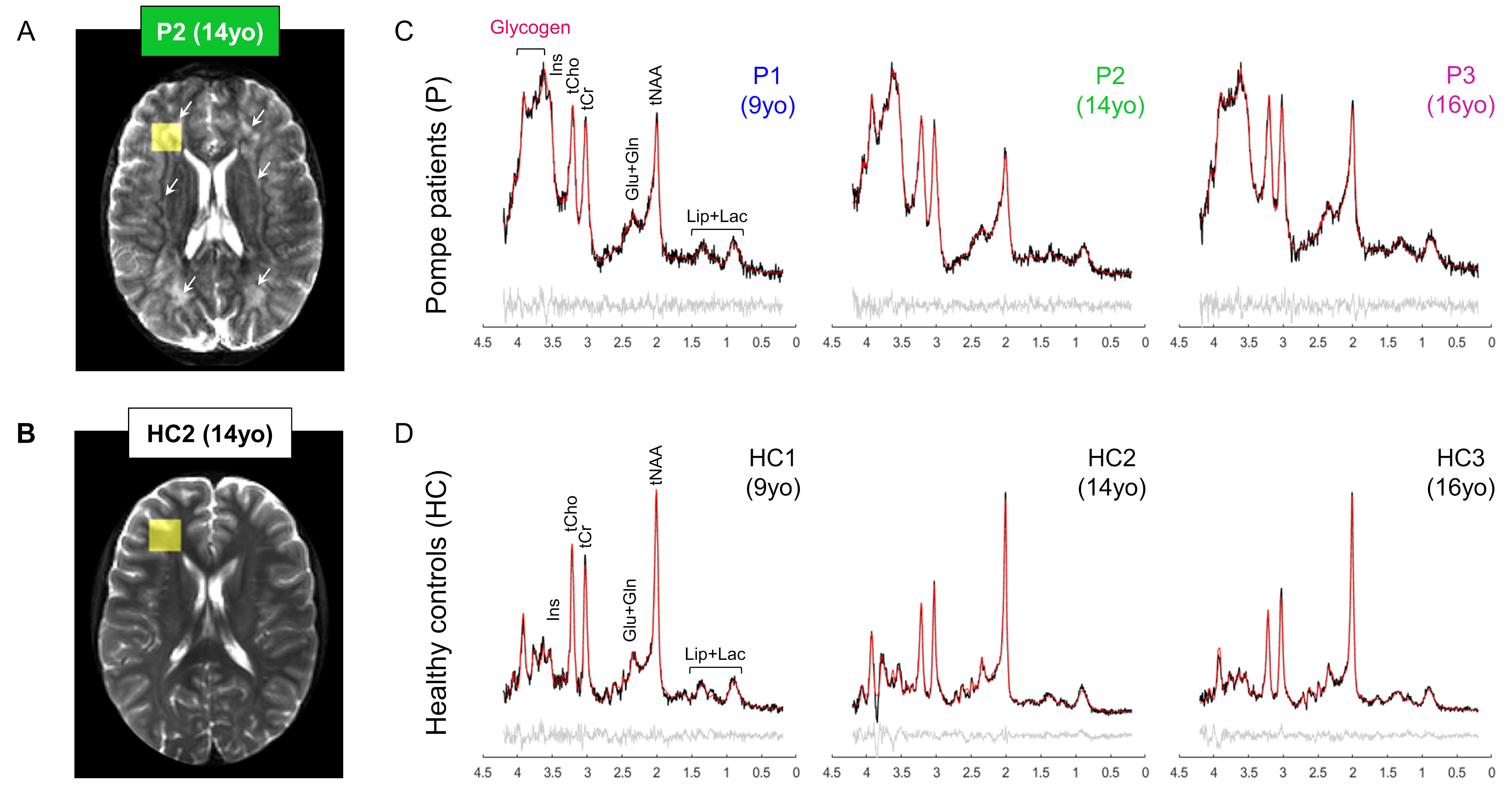
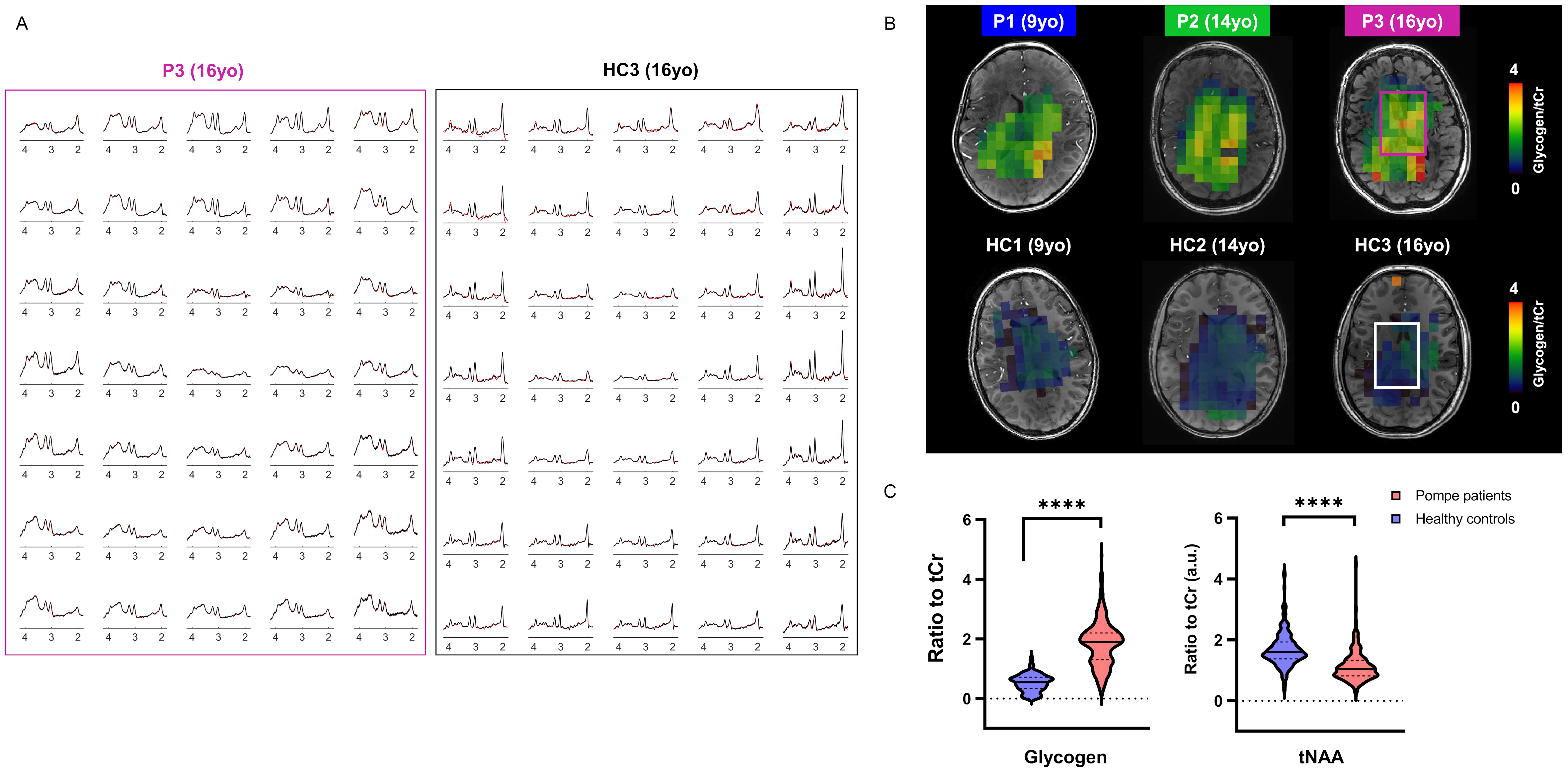
In vivo MR protocol and post-processing: A 3D-T1W gradient-echo (TR/TE=5.5/2.5ms, resolution 1x1x1mm3) and a multi-slice multi-echo (TR/TE1/TE2=4385/27/100ms, resolution 1.3x1.3x4mm3) acquisitions were performed and used for positioning of the volume-of-interest (VOI). SVS data were acquired using a sLASER sequence (TR/TE=6500/34ms, NSA=32, acq. time~5min) from a 18x18x18mm3 VOI positioned in the left frontal periventricular WM region (Fig.2B). For 2D-MRSI acquisition, volume pre-selection was performed using sLASER (TR/TE=5000/36-38ms, volume dimensions (AP,RL,FH)=130x90x10mm3, Fig.2B). Spatio-spectral encoding for the spectroscopic imaging was done using a concentric rings readout trajectory6 (FOV (AP,RL)=240x240mm, voxel size=10x10mm, slice thickness (FH)=10mm, acq. time~11min). FOCI refocusing pulses were used both for SVS and 2D-MRSI acquisitions to minimize in-plane chemical shift displacement errors. Outer-volume-suppression targeting lipid signal was performed using ten saturation bands interleaved with the water suppression and positioned circularly around the selected volume. SVS and 2D-MRSI data were reconstructed using in-house Matlab routines7 and spectra were fitted with LCModel8 with respective basis-sets.
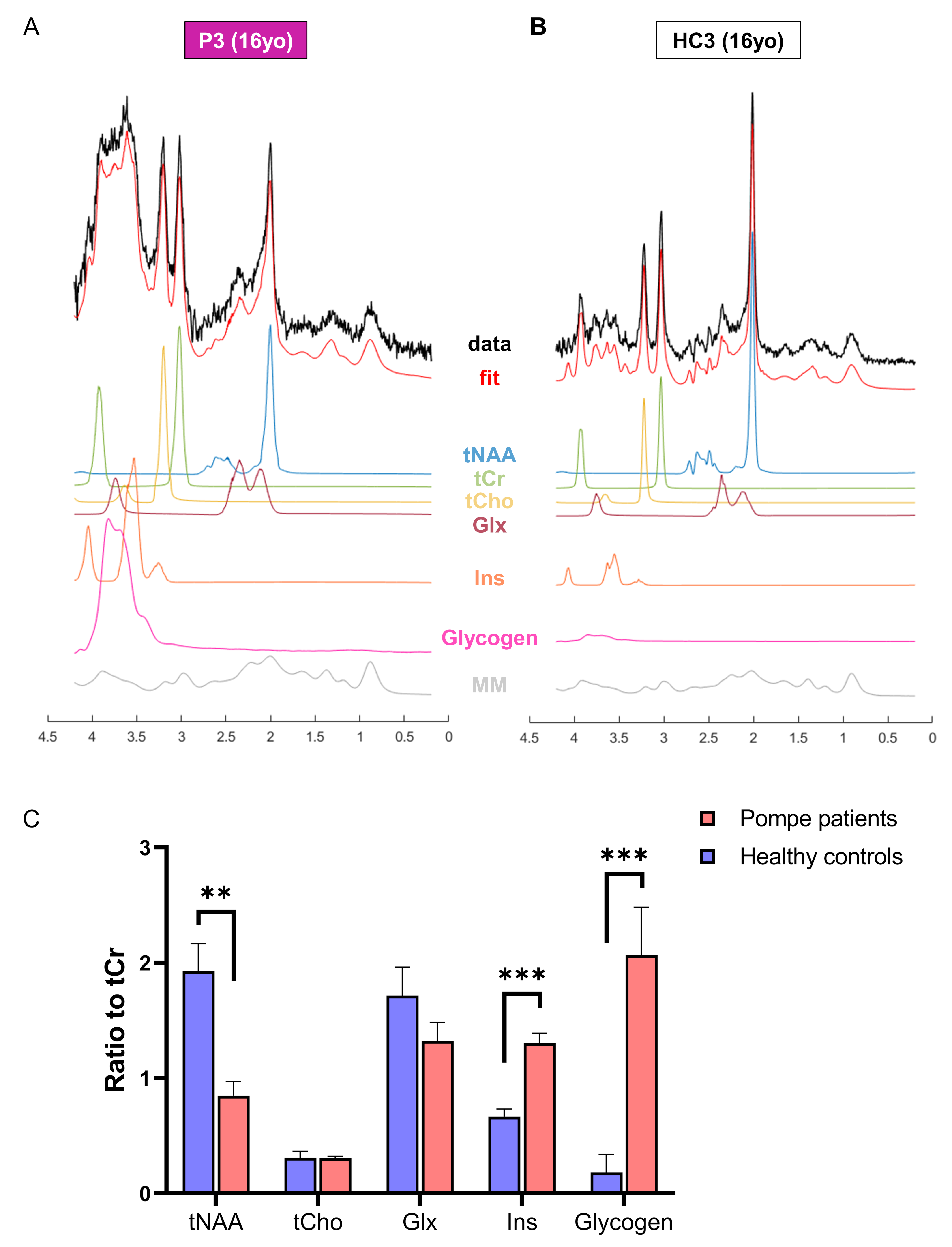
Glycogen phantom: A 28mM glycogen solution was prepared by mixing glycogen from rabbit liver (Sigma) with water. Data were acquired using same protocol as for SVS in vivo acquisition. The spectrum was corrected for frequency-drift and the residual water peak was removed with a linear prediction singular value decomposition Matlab-based routine. The resulting spectrum was added to the LCModel basis-sets to fit glycogen signal in vivo (Fig.4).
Statistical analysis: Statistical significance was tested using an unpaired Student’s t-test with Welch’s correction and assuming unequal variance (*p< 0.05, **p< 0.01, ***p< 0.001, GraphPad Software, USA).
Results and discussion
As illustrated in Fig.1&2, Pompe patients presented a decline in total intelligence and performance test scores, and showed widespread WM lesions on conventional T2-weighted images at time of the MRI. This is in agreement with previous reports3,4. Fig.3 shows SVS data acquired in Pompe patients (panels A/C) and healthy controls (panels B/D). A clear shift in the neurochemical profile is observed in all three patients, particularly, a large increase in signal between 3.5-4ppm attributed to glycogen, in agreement with expected glycogen accumulation in the brain of Pompe patients. The significant increase in glycogen was observed in combination with a significant increase in myo-inositol (Ins) and decrease in tNAA (NAA+NAAG) in patients compared to healthy controls (Fig.4A). As the signal of glycogen and Ins partially overlap at 3.6ppm (Fig.4A), quantification of Ins in patients is possibly compromised. The decrease in tNAA reflects neuronal damage in WM lesions and corroborates previous reports9. MRSI results were in line with the SVS data and showed a significant presence of glycogen as well as a significantly lower level of tNAA across the slice in patients (Fig.5). On the T2-weighted images, WM in the patients showed large hyperintense areas, possibly reflecting tissue damage associated with the glycogen accumulation seen on the MRSI.Conclusion
We illustrated the potential of MRS, both in SVS and in 2D-MRSI mode, to monitor the accumulation of glycogen in the brain of Pompe patients. As innovative treatment strategies which target both muscles and the brain are currently under development, MRS(I) could serve as a non-invasive readout to monitor disease progression and response to treatment. In addition, as neurochemical changes often precede macrostructural damages, MRS(I) might serve as an early biomarker for the effect of Pompe disease on the brain.Acknowledgements
This project has received funding from the Leiden University Fund (W-19356-2-32), and the H2020-MSCA-COFUND-LEaDing Fellow Programme (The Netherlands) under the Marie Skłodowska-Curie grant agreement No 707404.References
[1] van der Ploeg A.T. et al., Lancet (2008); [2] Van den Hout J.M.P. et al., Lancet (2000); [3] Ebbink B.J. et al., Neurology (2016); [4] Ebbink B.J. et al., Dev Med Child Neurol (2008); [5] Dasouki M. et al., Neurol Clin (2014); [6] Emir U.E. et al., NMR Biomed (2017); [7] Magnusson P.O. et al., Magn Reson Med (2019); [8] Provencher S.W., Magn Reson Med (1993); [9] Chien Y.H. et al., Pediatr Res (2006)
Figures