Idiopathic Lung Disease & Cystic Fibrosis
Sean B. Fain1
1University of Wisconsin-Madison, Madison, WI, United States
1University of Wisconsin-Madison, Madison, WI, United States
Synopsis
Idiopathic pulmonary fibrosis (IPF) and cystic fibrosis are deadly chronic lung diseases that have recently had breakthroughs in targeted drug therapies. Image-based biomarkers of ventilation and gas exchange using hyperpolarized 129Xe MRI are well suited to sensitively identify patients with progressive disease and monitor response to these new drug therapies. Additionally, ultra-short echo time (UTE) MRI has made significant advances in visualizing clinically relevant disease structures in both IPF and CF and can complement HP 129Xe MRI for longitudinal studies without requiring ionizing radiation. This structure-function imaging capability of MRI makes earlier and more frequent disease assessment feasible.
Introduction: Magnetic Resonance Imaging in Lung Disease
Chest X-ray and computed tomography are the modalities of choice for clinical imaging of lung disease. Magnetic resonance imaging is generally less utilized for lung diseases in the clinical setting but is increasingly used in clinical research. Continued improvement in ultra-short time to echo (UTE) [1, 2] and hyperpolarized 129Xe gas methods [3] and efficient sampling trajectories are driving increasing use in disease settings where non-ionizing radiation is advantageous, especially for longitudinal assessment of disease and pediatric lung diseases.As functional lung MRI becomes increasingly used for clinical research, a deeper understanding of the physiology of the lung becomes relevant. Hyperpolarized 129Xe MRI is essentially a functional imaging approach with repeatable quantitative analysis methods [4-6]. The distribution of hyperpolarized 129Xe MRI after inhalation acts as a gas tracer to provide a regional picture of gas distribution that is proportional to local lung ventilation [3]. Importantly, xenon is a soluble gas with different chemical shifts depending on gas, tissue/plasma, and red blood cell (RBC) local chemical environments [3], making it feasible to image regional xenon gas exchange.
Pulmonary Function: Restrictive vs. obstructive disease
These contrast mechanisms provide information about both restrictive and obstructive lung diseases. Restrictive lung disease refers to the decreased mechanical compliance due to fibrotic injury making it more difficult to inflate the lungs. Lung compliance can be calculated by dividing lung volume by inflation pressure. Spirometry is a clinical measure of the rate and cumulative volume of air flow at the mouth during a forced exhalation maneuver. Reduced forced vital capacity (FVC), which is the total volume of air that can be exhaled from total lung capacity, is a spirometric pulmonary function test indicative of decreased lung compliance in restrictive disease such as in interstitial lung disease (ILD), such as sarcoid and IPF.Obstructive lung disease is fundamentally different from restrictive lung disease and refers to blockage or narrowing of the airways such as in asthma and cystic fibrosis (CF). Reduced forced expiratory volume in 1 second (FEV1) is a spirometric pulmonary function test that is decreased in obstructive lung disease. Spirometric tests, though widely available and inexpensive, are fundamentally “backward-looking” in that they are insensitive to early disease processes and may not be significantly reduced until disease is quite progressed and less likely to be reversed by treatment interventions [7].
Clinical Relevance and Lung Disease Processes
Two important restrictive and obstructive lung diseases are interstitial lung disease (ILD) and cystic fibrosis (CF), respectively. Interstitial lung disease (ILD) is a broad class of fibrotic lung diseases that include chronic injury of the lungs due to aberrant inflammation, injury, and repair that contributes over time to vascular and airway injury [8]. The consequence is eventual damage that inhibits gas transfer and mechanically restricts ventilation due to reduced lung compliance. The “Idiopathic” component in the title of this talk refers to diseases of unknown cause, most commonly referring to idiopathic pulmonary fibrosis, or IPF. IPF makes up approximately 20-30% of ILD [9]; with a worldwide prevalence of approximately 3 million people. The incidence of IPF increases with age (typically >50 years), is most prominent in males and is one of the most difficult to treat of ILD lung diseases with typically poor outcomes. The diagnosis of IPF relies on a multi-disciplinary approach with a prominent component dependent on the pattern of lung fibrosis on chest CT.Cystic fibrosis (CF) is a rare (1:3,000-4,000 live births) but fatal genetic disorder caused by one of many mutations to the “Cystic fibrosis transmembrane conductance regulator (CFTR)” protein that conducts chloride ions across epithelial cell membranes. Most prominently, the CFTR mutations eventually cause chronic obstructive disease in the lungs but also affect the pancreas gastrointestinal tract, liver and male reproductive tract [10]. Of note, while the recessive disease is itself rare, with a world-wide prevalence of approximately 70,000, more than 10 million Americans are carriers of a CFTR gene mutation [11, 12]. CF in the lungs manifests early in young children as chronic infection due to the inability to move sticky, dehydrated mucus secretions from the airways because of impaired chloride channel function resulting in airway osmotic imbalance. Primarily due to impaired lung function, CF has until recently led to poor outcomes usually leading to death or transplant by the late twenties or early middle age in the best outcomes.
Role of Chest CT Imaging in Diagnosis and Characterizing Severity
The most common IPF pattern on chest CT (as distinguished from other forms of ILD) is called the usual interstitial pneumonia (UIP) pattern, which consists of specific textures on chest CT that correspond to characteristic structural features of inflammation and fibrosis. These include honeycombing, which appears as clusters of cystic spaces, typically in the posterior and base of the lung; reticulation, which appears as a network of fine lines; traction bronchiectasis, which appears as enlarged airways as lung parenchymal compliance decreases; and ground glass opacity, which is typically an indication of interstitial inflammation but is usually mixed with the other features in IPF [13]. However, chest CT is generally poor at predicting patient-specific progression, although advances in quantitative texture analysis show promise [14].In CF, chest CT is most common used to evaluate early obstructive disease severity and progression through evaluation of airway bronchiectasis, mucus plugging and air trapping using systematic scoring [15], with automated scoring systems recently advanced [16]. Airway bronchiectasis manifests due to chronic infection and thickened inflamed airways, often accompanied by central and distal airway plugging that leads to persistent obstruction and regional air trapping, indicated by low attenuation areas of lung parenchyma. However, even with the development of low X-ray dose CT protocols, the concerns about early and frequent CT imaging in childhood for longitudinal monitoring of CF disease progression persist for patients and parents. Consequently, MRI advances in this disease make it a promising and increasingly used method to both assess severity and therapy response.
Role for MRI in Monitoring Disease and Therapy
The common thread connecting these chronic lung diseases is that recent advances have led to FDA approved small molecule therapies that are promising treatment options for both CF [12], and IPF [7, 17]. These drugs appear to mitigate progression and have the potentially to reverse disease in recoverable lung regions. For IPF, the availability of two pharmacotherapeutic agents, pirfenidone and nintedanib, have been developed and clinical trials indicate they decrease physiological progression and likely improve progression-free survival. Current efforts are directed at identifying IPF early so that these therapies can have greater impact on patient outcomes, potentially relying on image biomarkers to assess therapy response [7]. The development of UTE MRI in concert with pulmonary perfusion [18, 19] and hyperpolarized 129Xe MRI provide a structure-function assessment with advantages for monitoring therapy response in IPF. The functional imaging of xenon transfer into the blood, is a strong candidate for a monitoring biomarker to detection progression and therapy response in IPF. Advances in hyperpolarized 129Xe spectroscopic imaging have begun to establish the ratio of the RBC-to-tissue/plasma (i.e. RBC:Barrier) signals, as a repeatable [6] and sensitive biomarker of IPF progression [20, 21] and pirfenidone and nintedanib treatment [22]. Moreover, emerging results suggest changes in the volume of well-ventilated lung and RBC:Barrier can detect functional abnormalities that are localized to non-fibrotic lung parenchyma on CT, i.e. regions not yet visually effected on chest CT.For CF, Ivacaftor and lumacaftor targeted to the most common CFTR mutations, and more recently, the triple combination therapy Trikafta (elexacaftor/ivacaftor/tezacaftor) that improve CFTR function and transport to the epithelial cell surface. Triple combination therapy shows promise for treating up to 90% of the CF population [23]. Early studies using hyperpolarized gas MRI demonstrated the strong regional response of ventilation to CFTR potentiation [24], and similar development of UTE MRI to evaluate disease severity in very young children provides a strong motivation for MRI as a comprehensive tool for structure-function assessment of CF lung disease that is safe for longitudinal evaluation in very young children [25, 26] and throughout the patient lifespan. Moreover, the burden of CF therapies is significant, and MRI can potentially play a role in personalized treatment approaches to further enhance patient quality of life [27].
Both diseases are likely at greater risk of severe COVID-19 lung disease. MRI may also play an important role in characterizing accelerated or persistent lung injury after SARS-CoV2 infection to phenotype patients who will benefit from interventions to mitigate the long-term sequelae of SARS-CoV2 infection [28]. In fact, ILD may share certain features with pulmonary vascular injury that leads to severe COVID-19 and may explain persistent symptoms after resolution of infection and likely plays a role in so-called “long-hauler” COVID-19 in which symptoms persist after apparent resolution of disease [29]. In CF, the data regarding COVID-19 are sparse mostly due to the CF population practicing stringent safety to avoid infection [30].
Acknowledgements
Slides provided by Dr. Laura Walkup and Dr. Jason Woods, Cincinnati Children's Hospital; and Dr. Talissa Altes and Dr. Robbie Thomen, University of Virginia and University of Missouri.References
- Johnson KM, Fain SB, Schiebler ML, Nagle S. Optimized 3D ultrashort echo time pulmonary MRI. Magn Reson Med. Nov 2013;70(5):1241-50. doi:10.1002/mrm.245702.
- Zhu X, Chan M, Lustig M, Johnson KM, Larson PEZ. Iterative motion-compensation reconstruction ultra-short TE (iMoCo UTE) for high-resolution free-breathing pulmonary MRI. Magn Reson Med. Apr 2020;83(4):1208-1221. doi:10.1002/mrm.279983.
- Ebner L, Kammerman J, Driehuys B, Schiebler ML, Cadman RV, Fain SB. The role of hyperpolarized (129)xenon in MR imaging of pulmonary function. Eur J Radiol. Jan 2017;86:343-352. doi:10.1016/j.ejrad.2016.09.0154.
- Zha W, Niles DJ, Kruger SJ, et al. Semiautomated Ventilation Defect Quantification in Exercise-induced Bronchoconstriction Using Hyperpolarized Helium-3 Magnetic Resonance Imaging: A Repeatability Study. Acad Radiol. Sep 2016;23(9):1104-14. doi:10.1016/j.acra.2016.04.0055.
- He M, Zha W, Tan F, Rankine L, Fain S, Driehuys B. A Comparison of Two Hyperpolarized (129)Xe MRI Ventilation Quantification Pipelines: The Effect of Signal to Noise Ratio. Acad Radiol. Jul 2019;26(7):949-959. doi:10.1016/j.acra.2018.08.0156.
- Hahn AD, Kammerman J, Evans M, et al. Repeatability of regional pulmonary functional metrics of Hyperpolarized (129) Xe dissolved-phase MRI. J Magn Reson Imaging. Oct 2019;50(4):1182-1190. doi:10.1002/jmri.267457.
- Martinez FJ, Collard HR, Pardo A, et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers. Oct 20 2017;3:17074. doi:10.1038/nrdp.2017.748.
- Kalchiem-Dekel O, Galvin JR, Burke AP, Atamas SP, Todd NW. Interstitial Lung Disease and Pulmonary Fibrosis: A Practical Approach for General Medicine Physicians with Focus on the Medical History. J Clin Med. Nov 24 2018;7(12)doi:10.3390/jcm71204769.
- Ryu JH, Moua T, Daniels CE, et al. Idiopathic pulmonary fibrosis: evolving concepts. Mayo Clin Proc. Aug 2014;89(8):1130-42. doi:10.1016/j.mayocp.2014.03.01610.
- Spielberg DR, Clancy JP. Cystic Fibrosis and Its Management Through Established and Emerging Therapies. Annu Rev Genomics Hum Genet. Aug 31 2016;17:155-75. doi:10.1146/annurev-genom-090314-05002411.
- Center for Disease Control and Prevention. Cystic Fibrosis. April 21, 2021, 2021. https://www.cdc.gov/genomics/disease/cystic_fibrosis.htm12.
- Ratjen F, Hug C, Marigowda G, et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir Med. Jul 2017;5(7):557-567. doi:10.1016/S2213-2600(17)30215-113.
- Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. Feb 2018;6(2):138-153. doi:10.1016/S2213-2600(17)30433-214.
- Fain SB, Lynch DA, Hatt C. Invited Commentary on "Quantitative CT Analysis of Diffuse Lung Disease". Radiographics. Mar-Apr 2020;40(2):E1-E3. doi:10.1148/rg.202020000515.
- Brody AS, Klein JS, Molina PL, Quan J, Bean JA, Wilmott RW. High-resolution computed tomography in young patients with cystic fibrosis: distribution of abnormalities and correlation with pulmonary function tests. J Pediatr. Jul 2004;145(1):32-8. doi:10.1016/j.jpeds.2004.02.03816.
- Perez-Rovira A, Kuo W, Petersen J, Tiddens HA, de Bruijne M. Automatic airway-artery analysis on lung CT to quantify airway wall thickening and bronchiectasis. Med Phys. Oct 2016;43(10):5736. doi:10.1118/1.496321417.
- Karimi-Shah BA, Chowdhury BA. Forced vital capacity in idiopathic pulmonary fibrosis--FDA review of pirfenidone and nintedanib. N Engl J Med. Mar 26 2015;372(13):1189-91. doi:10.1056/NEJMp150052618.
- Unites States Food and Drug Administration. FDA approves new breakthrough therapy for cystic fibrosis. Updated Oct. 21, 2019. Accessed April 21, 2021. https://www.fda.gov/news-events/press-announcements/fda-approves-new-breakthrough-therapy-cystic-fibrosis19.
- Altes TA, Johnson M, Fidler M, et al. Use of hyperpolarized helium-3 MRI to assess response to ivacaftor treatment in patients with cystic fibrosis. J Cyst Fibros. Mar 2017;16(2):267-274. doi:10.1016/j.jcf.2016.12.00420.
- Altes TA, Meyer CH, Mata JF, et al. Hyperpolarized helium-3 magnetic resonance lung imaging of non-sedated infants and young children: a proof-of-concept study. Clin Imaging. Sep - Oct 2017;45:105-110. doi:10.1016/j.clinimag.2017.04.00421.
- Roach DJ, Cremillieux Y, Fleck RJ, et al. Ultrashort Echo-Time Magnetic Resonance Imaging Is a Sensitive Method for the Evaluation of Early Cystic Fibrosis Lung Disease. Ann Am Thorac Soc. Nov 2016;13(11):1923-1931. doi:10.1513/AnnalsATS.201603-203OC22.
- Woods JC, Wild JM, Wielputz MO, et al. Current state of the art MRI for the longitudinal assessment of cystic fibrosis. J Magn Reson Imaging. Nov 2020;52(5):1306-1320. doi:10.1002/jmri.2703023.
- Wang K, Schiebler ML, Francois CJ, et al. Pulmonary perfusion MRI using interleaved variable density sampling and HighlY constrained cartesian reconstruction (HYCR). J Magn Reson Imaging. Sep 2013;38(3):751-6. doi:10.1002/jmri.2401824.
- Torres L, Kammerman J, Hahn AD, et al. "Structure-Function Imaging of Lung Disease Using Ultrashort Echo Time MRI". Acad Radiol. Mar 2019;26(3):431-441. doi:10.1016/j.acra.2018.12.00725.
- Carey KJ, Hahn A, Torres LA, et al. Quantitative Magnetic Resonance Imaging and Computed Tomography Measures of Progression in Idiopathic Pulmonary Fibrosis. American Journal of Respiratory and Critical Care Medicine. 2020;201:A4510.26.
- Hahn A, Carey KJ, Barton GP, et al. Functional MRI of Regional Gas Exchange in IPF Disease Progression. American Journal of Respiratory and Critical Care Medicine. 2020;201:A5995.27.
- Hahn A, Carey K, Sandbo N, et al. Response of Hyperpolarized 129Xe MRI measures of ventilation and gas-exchange to anti-fibrotic treatment in Idiopathic Pulmonary Fibrosis. 2021:28.
- Myall KJ, Mukherjee B, Castanheira AM, et al. Persistent Post-COVID-19 Inflammatory Interstitial Lung Disease: An Observational Study of Corticosteroid Treatment. Ann Am Thorac Soc. Jan 12 2021;doi:10.1513/AnnalsATS.202008-1002OC29.
- Colombo C, Burgel PR, Gartner S, et al. Impact of COVID-19 on people with cystic fibrosis. Lancet Respir Med. May 2020;8(5):e35-e36. doi:10.1016/S2213-2600(20)30177-630.
- Carfi A, Bernabei R, Landi F, Gemelli Against C-P-ACSG. Persistent Symptoms in Patients After Acute COVID-19. JAMA. Aug 11 2020;324(6):603-605. doi:10.1001/jama.2020.12603
Figures

The histopathology of IPF shows deposition of fibrotic tissue that thickens the septal wall of the alveolar sacs preventing effective gas exchange. Top panel at left: region of healthy lung with thin alveolar walls. Panel B: an alveolus with highly thickened septal walls. IPF makes up approximately 20-30% of ILD with mean survival of 2.5–5 years after definite diagnosis and greater prevalence in older males (>50 years). Recent advances in anti-fibrotic therapies interfere with fibrotic tissue deposition and show promise for mitigating progression.
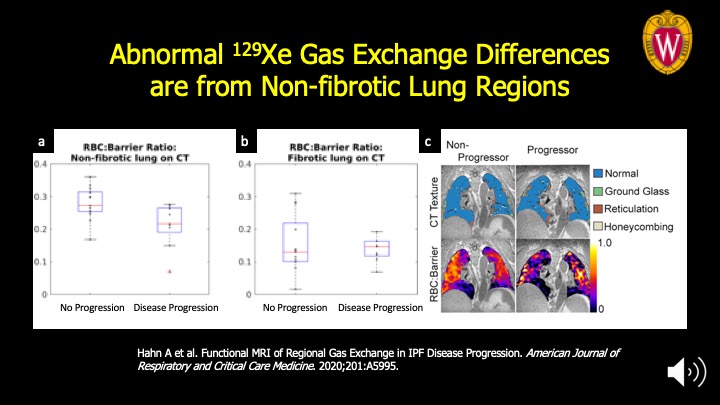
The signal from impaired gas exchange that distinguishes progressive from stable disease is coming from the non-fibrotic lung regions on CT imaging. This highlights the importance of functional imaging of gas exchange to detect regions where there may be potential for reversing disease. Panel A: the differences between the progression and stable sub-populations are significantly different at P<0.03 vs. the fibrotic lung regions in Panel B. Panel C: Mapping of the functional maps onto quantitative CT texture is seen at far right for representative patients.
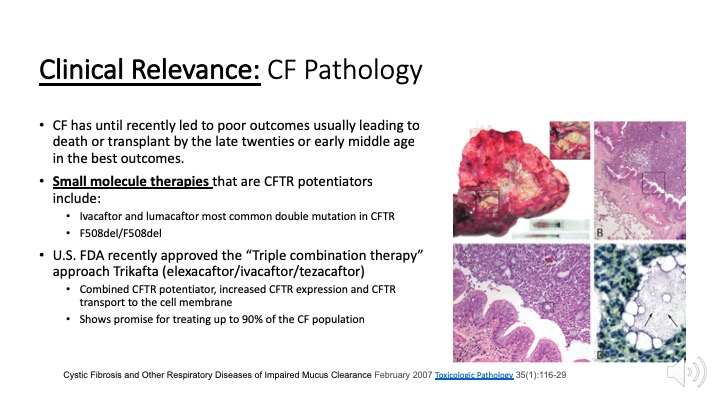
CF pathology is driven by mucus impaction in the airways that harbor infectious bacterial colonies eventually leading to chronic inflammation and airway remodeling (ROI) within this transplanted lung. Small areas of bacterial colonies are dispersed throughout and visible upon magnification. Recently developed drugs target to the CFTR protein to improve its function and increase expression on the airway epithelium. "Triple combination therapy” recently approved by the US FDA target a broader cross-section of up to 90% of CF patients.
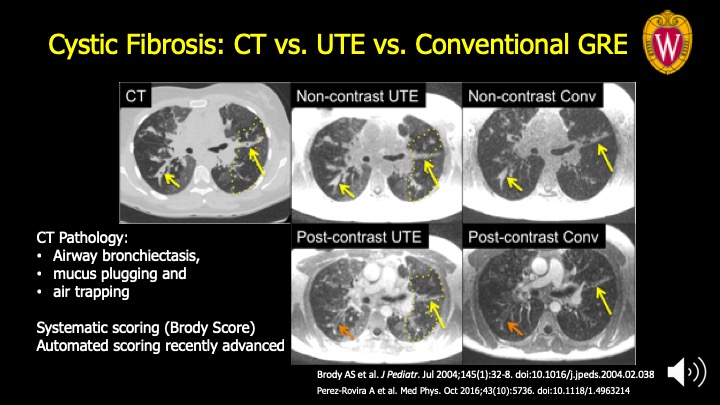
The chronic mucus plugging and airway injury manifests on structural imaging as bronchiectasis, opaque airway lumen due to plugged, and air trapping as seen on CT at left (dotted line). All these features are also seen on UTE MRI (middle) compared to chest CT as a reference at the left. These features make up the main components of the Brody score, a categorical scoring system to evaluate CF severity. Conventional GRE (far right) detects some of these features with variable confidence but not air trapping.