1200
AICAR prevents heart failure in a rat model of doxorubicin-induced cardiotoxicity1Department of Physiology, Anatomy and Genetics, University of Oxford, Oxford, United Kingdom, 2MRC Mitochondrial Biology Unit, University of Cambridge, Cambridge, United Kingdom
Synopsis
Doxorubicin (DOX) is a commonly used chemotherapeutic agent for the treatment of many cancers. However, DOX has serious cardiotoxic side effects culminating in congestive heart failure. We have previously shown in a clinically-relevant rat model of DOX-induced heart failure (DOX-HF), that this is due to loss and dysfunction of mitochondria. We show here that 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), an activator of AMPK, can prevent heart failure in DOX-treated rats. This cardioprotective effect appears to be, at least in part, achieved through improved fatty acid oxidation in cardiac mitochondria which can be indirectly assessed with hyperpolarized [2-13C]pyruvate MRS.
Introduction
Doxorubicin (DOX) is a commonly used anthracycline chemotherapeutic agent for the treatment of many cancers. However, DOX is cardiotoxic, causing heart failure in ~5% of patients1. Different mechanisms have been proposed to cause this cardiotoxicity, including mitochondrial impairment2. Hyperpolarized MRS by dissolution dynamic nuclear polarization of 13C-labelled substrates3 has revolutionized metabolic flux measurements in preclinical models4 and in the human heart5,6. We have previously shown in a clinically-relevant rat model of doxorubicin-induced heart failure (DOX-HF), that cardiac metabolic fluxes, assessed by hyperpolarized 13C magnetic resonance spectroscopy (MRS), are decreased in the hearts of DOX-treated rats (abstract 3605 ISMRM 2017) and that this is not due to oxidative stress but due to a loss and dysfunction of mitochondria (abstract 0786 ISMRM 2019). We now set out to establish whether 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), which activates AMPK7, increasing mitochondrial biogenesis8 and fatty acid oxidation9, can prevent heart failure in DOX-treated rats.Methods
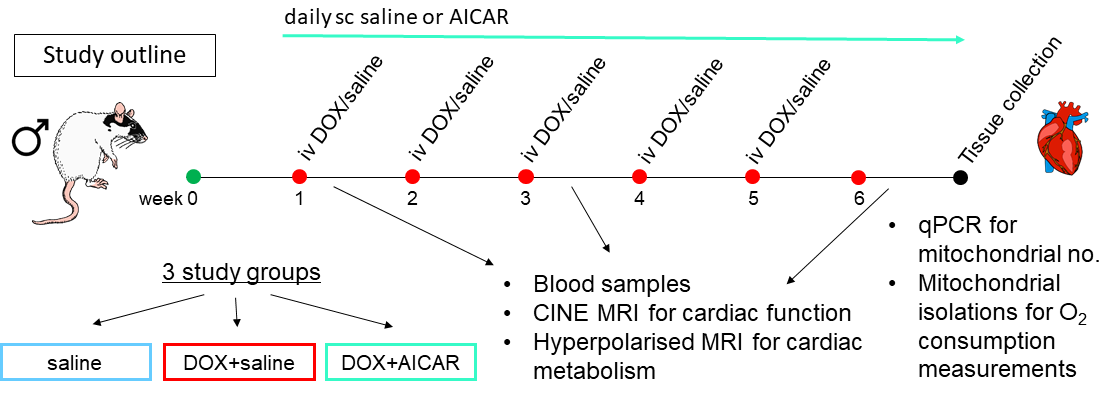
Male Wistar rats were weight-matched and divided into 3 groups (Fig 1): a saline control group (n=6) receiving weekly iv injections of saline for 5 weeks, a DOX+saline group (n=9) receiving weekly iv injections of DOX (3 mg/kg) for 5 weeks and daily sc injections of saline and a DOX+AICAR group (n=9) receiving weekly iv injections of DOX (3 mg/kg) for 5 weeks and daily sc injections of AICAR (500 mg/kg). Body weight was monitored weekly. At weeks 1, 3 and 6 of the study, cardiac function was assessed with a CINE-FLASH sequence using a 72-mm dual-tuned 1H/13C birdcage volume transmit and a four-channel 1H-receive coil (Rapid Biomedical), acquiring 8-10 cardiac-gated short axis slices (1.6 mm thick) of the heart. Images were analyzed using the free hand drawing function in ImageJ. [2-13C]pyruvate was hyperpolarized on a prototype hyperpolarizer (Oxford Instruments) for 45 min. One mL of 80 mM hyperpolarized [2-13C]pyruvate was injected into the tail vain over 10s and 13C MR spectra were acquired over the heart using a 13C two-channel surface receive coil (Rapid Biomedical) every second for 60s (10 mm slab, hard pulse, 13° flip angle, bandwidth 17kHz). Multi-coil spectra were added in phase, and the first 30s of spectra from the initial appearance of the pyruvate peak were summed and quantified with AMARES/jMRUI10. Blood samples were taken at all 3 time points and the plasma was analyzed for non-esterified fatty acid (NEFA) content using a spectrophotometric assay kit (Randox). At week 6, rats were sacrificed, hearts excised and subsarcolemmal (SSM) and interfibrillar (IFM) mitochondria were isolated for mitochondrial oxygen consumption measurements with a Clarke-type oxygen electrode as previously described11.Results
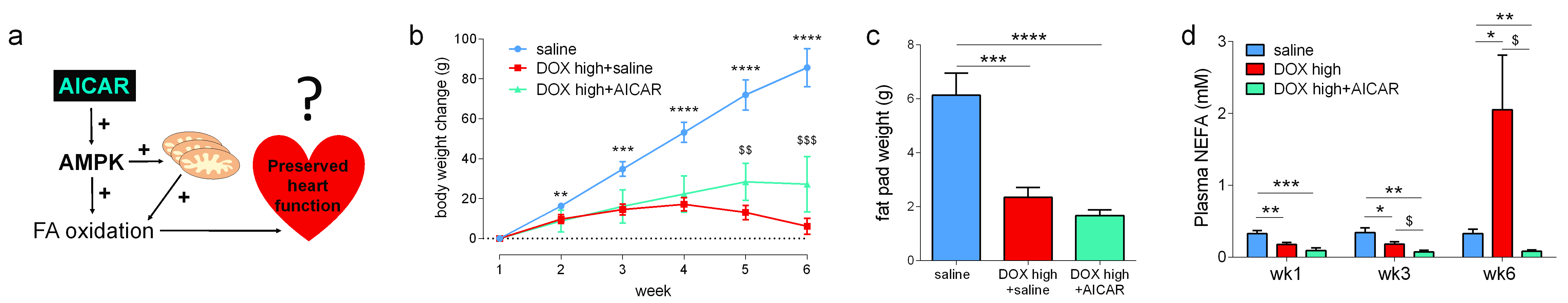
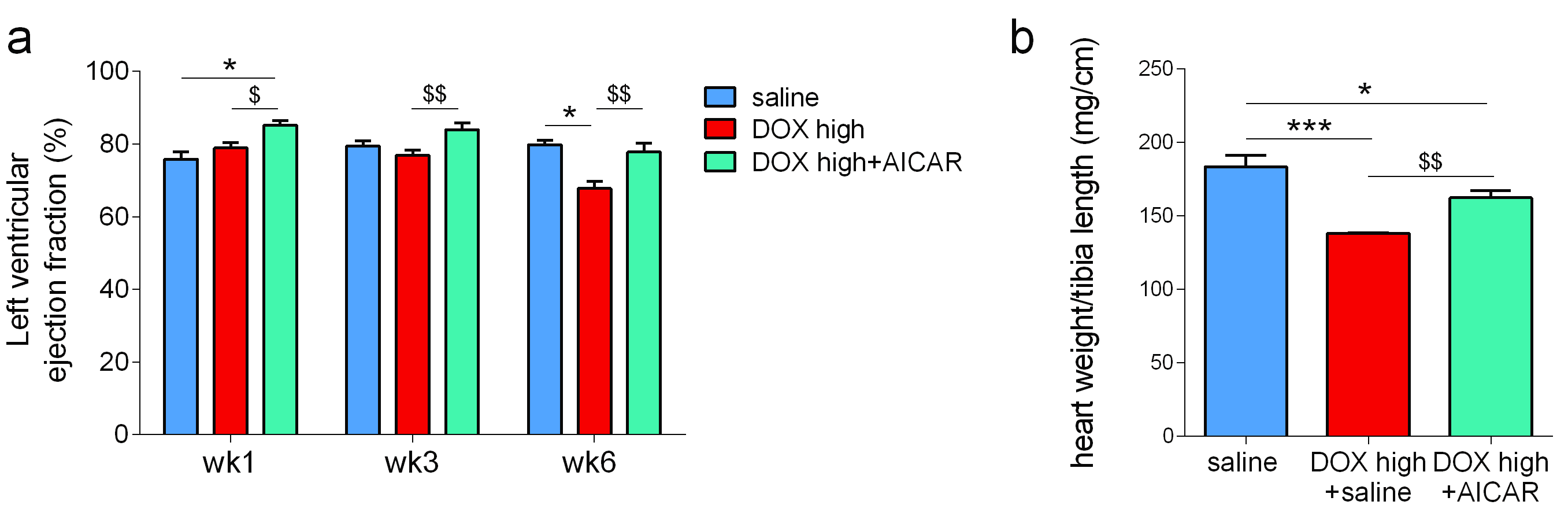
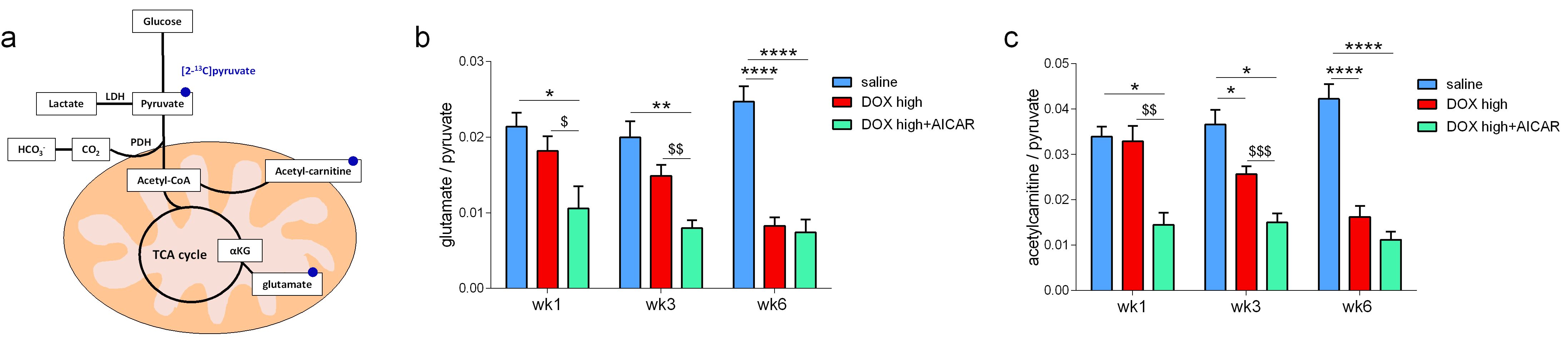
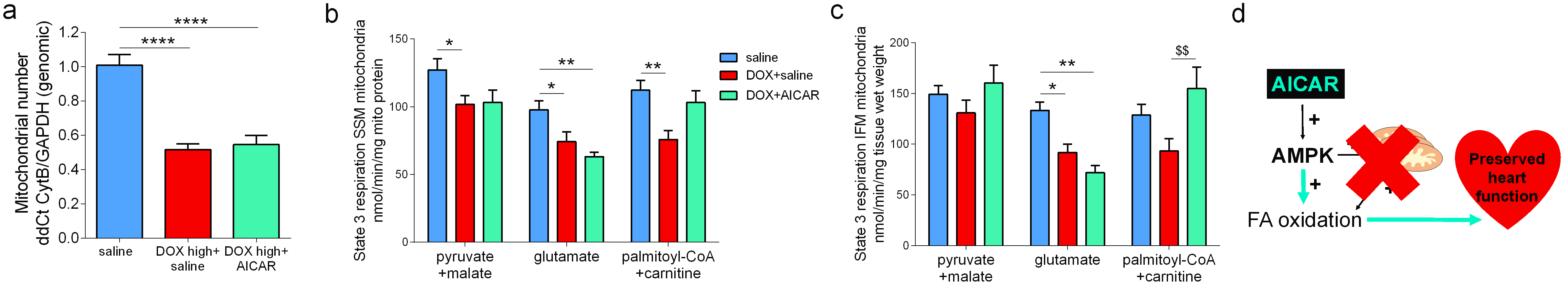
We hypothesized that AICAR could achieve cardioprotection in DOX-HF by increasing mitochondrial biogenesis and/or increasing mitochondrial fatty acid oxidation (Fig 2a). Both DOX+saline rats and DOX+AICAR rats gained significantly less weight over the study compared to saline control rats. However, DOX+AICAR rats showed significantly increased body weight gain compared to DOX+saline rats (Fig 2b). Epididymal fat pad weights in rats at the end of the study were reduced in both DOX+saline and DOX+AICAR rats compared to saline control rats (Fig 2c). AICAR normalized plasma NEFA levels in DOX-treated rats at week 6 of the study (Fig 2d). DOX+saline rats had a significantly reduced ejection fraction (EF) compared to saline treated control rats at week 6 and this decrease in EF was prevented in DOX+AICAR rats (Figure 3a). The heart weight:tibia length ratio, indicative of cardiac atrophy, was decreased in both DOX groups compared to saline controls at week 6 but was significantly higher in DOX+AICAR rats than DOX+saline rats (Fig 3b). We next assessed cardiac metabolic fluxes with hyperpolarized [2-13C]pyruvate MRS (Fig 4a). There was a marked decrease of tricarboxylic acid (TCA)-cycle derived glutamate labelling at week 6 in DOX+saline treated rats (Fig 4b) and of acetyl-carnitine labelling at weeks 3 and 6 (Fig 4c). Interestingly, both glutamate and acetyl-carnitine labeling were decreased in DOX+AICAR rats already at week 1 (Fig 4b-c). Mitochondrial number was decreased in both DOX+saline and DOX+AICAR rat compared to saline-treated controls (Fig 5a). SSM isolated from high dose DOX hearts showed a decreased oxygen consumption rate with all substrates (Fig 5b), while IFM had a reduced oxygen consumption rate only with glutamate (Fig 5c). Both SSM and IFM from DOX+AICAR rats showed reduced oxygen consumption rates only with glutamate. However, oxygen consumption was significantly improved in IFM with palmitoyl-CoA+carnitine (Fig 5c), suggesting that increased fatty acid oxidation may be the mechanism of AICAR-cardioprotection in this model of DOX-HF (Fig 5d).Discussion and conclusion
We have shown here that AICAR can prevent DOX-HF in rats. AICAR did not increase mitochondrial number in DOX-treated hearts, however, AICAR improved fatty acid oxidation in cardiac mitochondria from DOX-treated rats, which may overall allow improved cardiac energetics despite low mitochondrial number, as fatty acids are more energy efficient, producing more ATP per mole than glucose. Hyperpolarized [2-13C]pyruvate showed an early decrease in carbohydrate-derived TCA cycle flux in DOX+AICAR rats, which may reflect increased fatty acid oxidation due to reciprocal inhibition from the Randle cycle12. AICAR is already being used in clinical trials for other pathologies4 and its use in cancer patients receiving DOX is therefore feasible in the future to prevent DOX-HF.Acknowledgements
This work was supported by a British Heart Foundation Immediate Postdoctoral Basic Science Research Fellowship (FS/16/7/31843).References
1. Yu AF, Jones LW. Breast cancer treatment-associated cardiovascular toxicity and effects of exercise countermeasures. Cardio-Oncology. 2016. doi:10.1186/s40959-016-0011-5.
2. Varga Z V, Ferdinandy P, Liaudet L, Pacher P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am J Physiol - Hear Circ Physiol. 2015;309(9):H1453-H1467. doi:10.1152/ajpheart.00554.2015.
3. Ardenkjaer-Larsen JH, Fridlund B, Gram A, et al. Increase in signal-to-noise ratio of > 10,000 times in liquid-state NMR. Proc Natl Acad Sci U S A. 2003;100(18):10158-10163. doi:10.1073/pnas.1733835100.
4. Timm KN, Miller JJ, Henry JA, Tyler DJ. Cardiac applications of hyperpolarised magnetic resonance. Prog Nucl Magn Reson Spectrosc. 2018;106-107:66-87. doi:10.1016/j.pnmrs.2018.05.002.
5. Cunningham CH, Lau JYC, Chen AP, et al. Hyperpolarized 13C Metabolic MRI of the Human Heart: Initial Experience. Circ Res. 2016;119(11):1177-1182. doi:10.1161/CIRCRESAHA.116.309769.
6. Tyler D, Rider O, Dodd M, et al. Demonstrating the Randle Cycle in Vivo: Assessment of Physiological Alterations in Human Cardiac Metabolism Using Hyperpolarised 13C MR Spectroscopy. In: International Society for Magnetic Resonance in Medicine. Honolulu, Hi, USA; 2017:0726.
7. Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5‐Aminoimidazole‐4‐Carboxamide Ribonucleoside: A Specific Method for Activating AMP‐Activated Protein Kinase in Intact Cells? Eur J Biochem. 1995. doi:10.1111/j.1432-1033.1995.tb20498.x.
8. Kukidome D, Nishikawa T, Sonoda K, et al. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes. 2006. doi:10.2337/diabetes.55.01.06.db05-0943.
9. O’Neill HM, Lally JS, Galic S, et al. AMPK phosphorylation of ACC2 is required for skeletal muscle fatty acid oxidation and insulin sensitivity in mice. Diabetologia. 2014. doi:10.1007/s00125-014-3273-1.
10. Vanhamme L, Van Den Boogaart A, Van Huffel S. Improved Method for Accurate and Efficient Quantification of MRS Data with Use of Prior Knowledge. J Magn Reson. 1997;129(1):35-43. doi:10.1006/jmre.1997.1244.
11. Heather LC, Cole MA, Tan JJ, et al. Metabolic adaptation to chronic hypoxia in cardiac mitochondria. Basic Res Cardiol. 2012;107(3):268. doi:10.1007/s00395-012-0268-2.
12. Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Metab. 2009. doi:10.1152/ajpendo.00093.2009.
Figures