0899
Brain structure in the homozygous FUSDelta14 mouse recapitulates amyotrophic lateral sclerosis phenotypes1Wellcome Centre for Integrative Neuroimaging, FMRIB, Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, United Kingdom, 2Mouse Imaging Centre, The Hospital for Sick Children, Toronto, ON, Canada, 3Mammalian Genetics Unit, MRC Harwell Institute, Oxfordshire, United Kingdom, 4Department of Neuromuscular Diseases, Institute of Neurology, University College London, London, United Kingdom
Synopsis
This study assesses changes in brain anatomy with MRI in the homozygous humanized FUSDelta14 mouse model of amyotrophic lateral sclerosis (ALS). Post-mortem brain T2w-images were acquired at 7T, with 40μm isotropic resolution. After registration, the deformation fields were compared between mutant and wild-type mice. Homozygous FUSDelta14 mice exhibited atrophy in multiple grey and white matter structures. These results are in agreement with observations such as cortical thinning and alterations in white matter microstructure in ALS patients. Homozygous humanized FUSDelta14 mice show an early brain phenotype and are therefore a promising model for the study of ALS pathogenic mechanisms.
Introduction
Mutations in a subset of RNA binding proteins, including FUS, lead to aberrant RNA metabolism and are a relevant known cause of ALS1. The recently developed FUSDelta14 knock-in mouse model expresses physiological levels of a partially-humanized FUS gene harboring a human early-onset ALS truncation mutation. Heterozygous FUSDelta14 mice show progressive degeneration of motor neurons and loss of motor function, but the phenotype is detected only after 12 months of age2. Mutations in FUS are associated with a pathological gain-of-function cascade3, thus an accentuated and/or early phenotype is expected in homozygous FUSDelta14 mice. In this study, we used MRI to assess neuroanatomical alterations in 3-month-old homozygous FUSDelta14 mice.Materials and Methods
Nine homozygous FUSDelta14 mice and 10 wild-type (WT) littermates (females, 10-12 weeks-old) were studied. Mice were deeply anesthetized with ketamine/xylazine and intracardially perfused with a first flush of saline solution, followed by formalin 4%. Both saline and formalin solutions contained 2mM Gd-contrast agent (Gd-CA; Gadovist, Bayer Vital GmbH, Leverkusen, Germany). Heads were removed and skulls were dissected from the skin, lower jaw, muscles and ears. Brains were kept in the skull, immersed in 4% formalin with 2mM Gd-CA for 48h, and then kept in PBS with 2mM Gd-CA and 0.05% azide until scanned. MRI was performed on a 7.0 tesla MRI scanner (Agilent Inc., Palo Alto, CA). Sixteen custom-built solenoid coils were used to image the brains in parallel4. Anatomical images with 40μm isotropic resolution were acquired using a T2-weighted 3D fast spin-echo sequence, with TR=350 ms, TE=n*12ms, n=1,…6 echoes (TEeff=30ms), field-of-view of 20 x 20 x 25 mm3, matrix size = 504 x 504 x 630, 4 averages and cylindrical acquisition of k-space. Total imaging time was approximately 14 hours5. Brain images were registered to a consensus space. The deformation field necessary to bring each brain to this space was used to compute voxel volumes, represented as the logarithm of the Jacobian determinant. Volumes were modelled firstly as a function of genotype, and secondly as a function of both genotype and mouse weight to account for variations in the animal size. Results are presented after false discovery rate (FDR) multiple comparison correction.Results
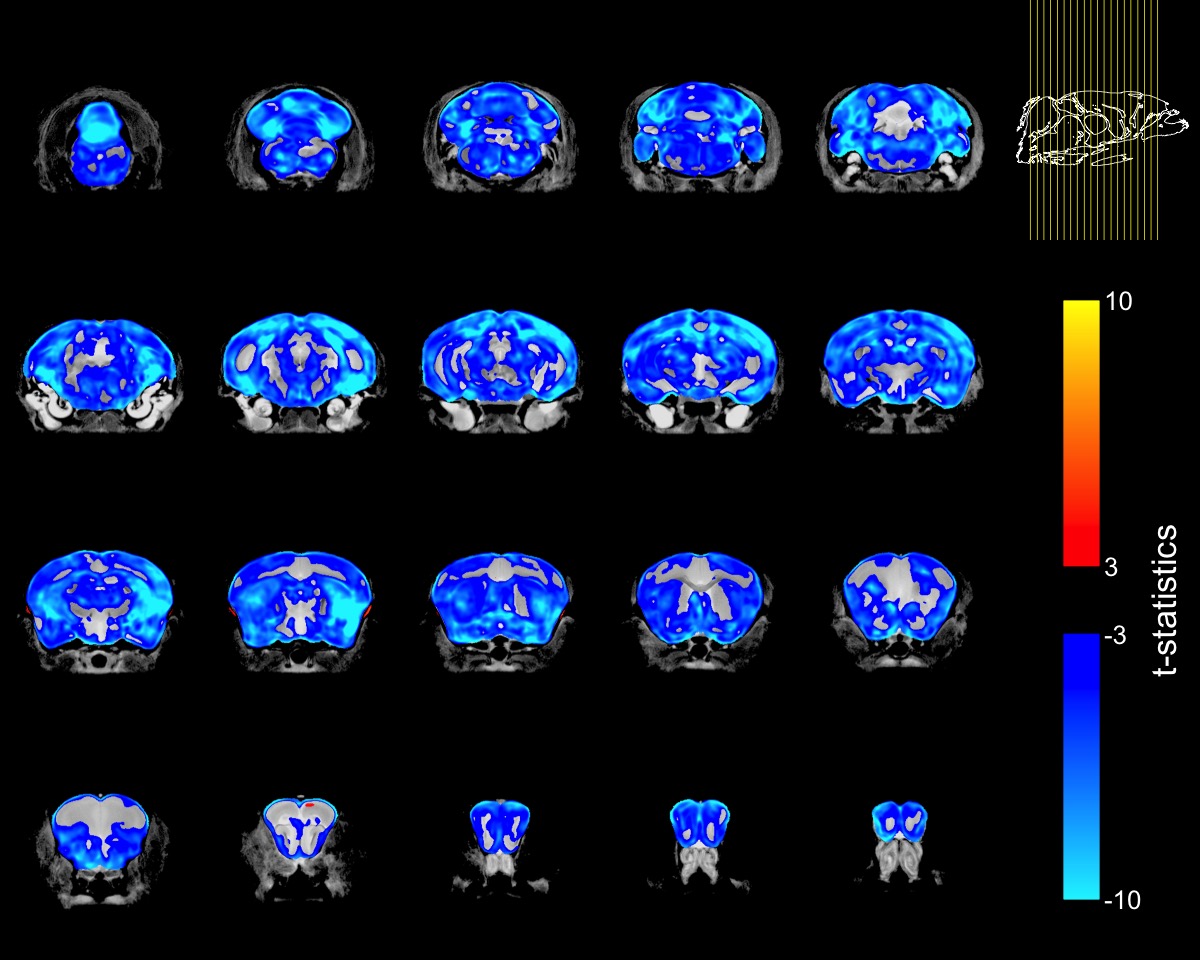
Homozygous FUSDelta14 mice were approximately 20% smaller than WT littermates (FUSDelta14: 19.1±1.2 g, WT: 23.6±1.9 g, p<0.0001), and had a 15% smaller brain volume (FUSDelta14: 373.6±12.8 mm3, WT 439.2±9.7 mm3, p<0.0001). Brain volume and terminal weight were highly correlated (R=0.92, R2=0.83, p<0.001).When the voxel-wise Jacobian determinants were modeled as a function of genotype only, widespread brain atrophy was observed (Figure-1). Only a few regions did not show reduction in volume, including subregions in the cingulate cortex, the medial preoptic nucleus, granule cell layers in the olfactory and accessory olfactory bulbs, the fourth ventricle, cerebral aqueduct and periaqueductal grey matter. All other regions in the cerebrum, cerebellum and brainstem were smaller in FUSDelta14 mice when only genotype was considered as an explanatory variable and at an FDR of 1%.
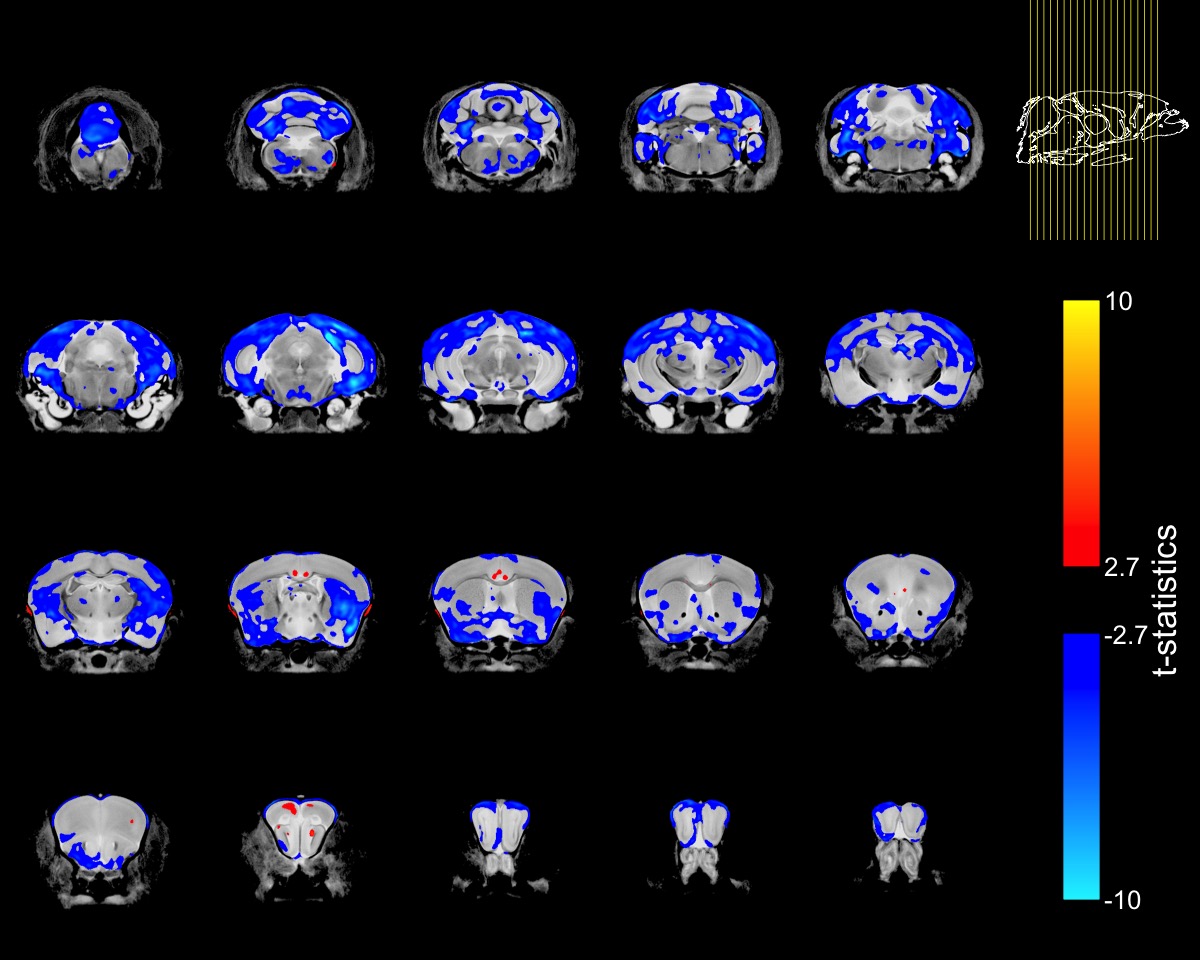
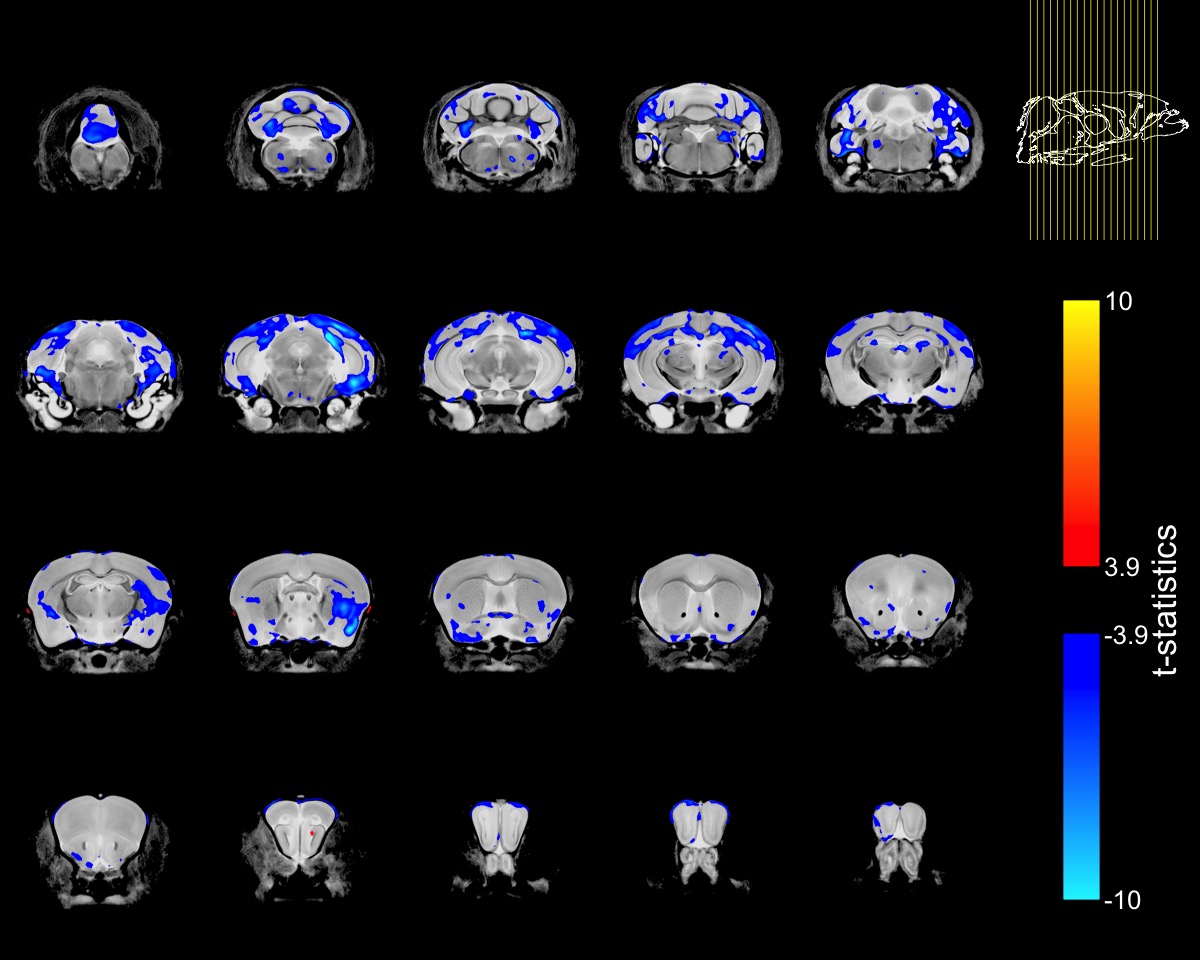
To account for the smaller size of FUSDelta14 mice, the Jacobian determinants were also modeled as a function of genotype and weight. The condition number of the matrix containing genotype and weight for each mouse was 39.6, indicating some degree of collinearity but a relatively stable model estimation. In this combined analysis, genotype had a significant effect in modelling the Jacobian determinants at FDRs of 5% and 1%. Figure-2 shows in blue the atrophic regions in FUSDelta14 mice when considering weight as a covariate and with an FDR of 5%, while in Figure-3 these results are presented with an FDR of 1%. Most of the observed spatial patterns in t-statistics maps are bilaterally symmetric (Figures 1-3).
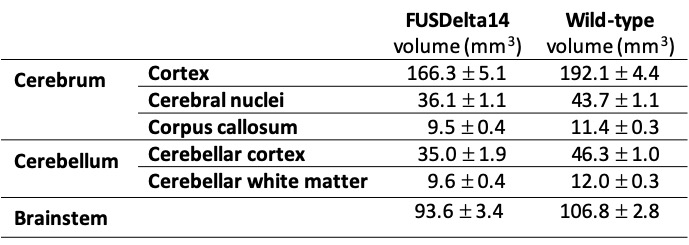
The structures with higher t-statistics were located in the cerebellum, with atrophy in multiple white and grey matter structures at FDR 1%. In the cerebrum, atrophy was observed in different cortical regions, including the secondary (FDR of 1%) and the primary (FDR 5%) motor cortices. At FDR 1%, the anterior commissure and the corpus callosum were also atrophic in FUSDelta14 mice, as well as hippocampus, striatum and olfactory bulbs. Table-1 shows volumes in different brain structures for FUSDelta14 and wild-type mice.
Discussion
These findings recapitulate pathological features described in ALS patients, such as cortical thinning6, reduced cerebellar grey matter volume7 and atrophy of subcortical structures8. Altered fractional anisotropy, radial and axial diffusivity have also been observed in white matter of ALS patients7, pointing to a spread involvement of brain white matter structures in the disease. Similarly, reduced axonal organization has been hypothesized from altered diffusion MRI metrics in the corpus callosum, cortex and hippocampus of SOD1-G93A transgenic mice, a different model of ALS9.Here we have shown that homozygous FUSDelta14 mice present anatomical alterations in the brain, in agreement with observations in ALS patients and in SOD1 mice. These anatomical anomalies were present much earlier than the first alterations observed in heterozygous FUSDelta14 mice.
Conclusions
Homozygous humanized FUSDelta14 mice recapitulate brain anatomical features observed in ALS patients, with atrophy of grey and white matter structures, and with an early phenotype presentation, being thus a valuable model to study the disease mechanisms in ALS.Acknowledgements
The authors Jason Lerch and Karla L. Miller contributed equally to this study.
This work was supported by the Wellcome Trust (grant 202788/Z/16/Z), MRC and Harwell funding. The Wellcome Centre for Integrative Neuroimaging is supported by core funding from the Wellcome Trust (grant 203139/Z/16/Z).
References
1. Vance C, Rogelj B, Hortobagyi T, et al. Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6. Science. 2009;323(5918):1208-1211
2. Devoy A, Kalmar B, Stewart M, et al. Humanized mutant FUS drives progressive motor neuron degeneration without aggregation in “FUSDelta14” knockin mice. Brain. 2017;140(11):2797-2805.
3. Shiihashi G, Ito D, Yagi T, Nihei Y, Ebine T, Suzuki N. Mislocated FUS is sufficient for gain-of-toxic-function amyotrophic lateral sclerosis phenotypes in mice. Brain. 2016;139(9):2380-2394.
4. Bock NA, Nieman BJ, Bishop JB, Henkelman RM. In vivo multiple-mouse MRI at 7 Tesla. Magn Reson Med. 2005;54(5):1311-1316.
5. Spencer Noakes TL, Henkelman RM, Nieman BJ. Partitioning k -space for cylindrical three-dimensional rapid acquisition with relaxation enhancement imaging in the mouse brain. NMR Biomed. 2017;30(11):e3802.
6. Verstraete E, Veldink JH, Hendrikse J, Schelhaas HJ, van den Heuvel MP, van den Berg LH. Structural MRI reveals cortical thinning in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2012;83(4):383-388.
7. Christidi F, Karavasilis E, Riederer F, et al. Gray matter and white matter changes in non-demented amyotrophic lateral sclerosis patients with or without cognitive impairment: A combined voxel-based morphometry and tract-based spatial statistics whole-brain analysis. Brain Imaging Behav. 2018;12(2):547-563.
8. Westeneng H-J, Verstraete E, Walhout R, et al. Subcortical structures in amyotrophic lateral sclerosis. Neurobiol Aging. 2015;36(2):1075-1082.
9. Gatto RG, Amin M, Finkielsztein A, et al. Unveiling early cortical and subcortical neuronal degeneration in ALS mice by ultra-high field diffusion MRI. Amyotroph Lateral Scler Front Degener. 2019;20(7-8):549-561.
Figures