0636
Bright-Ferritin: A novel MRI gene reporter complex for sensitive and longitudinal cell tracking1Institute of Biomaterials and Biomedical Engineering, University of Toronto, Toronto, ON, Canada, 2Translational Biology & Engineering Program, Ted Rogers Centre for Heart Research, Toronto, ON, Canada, 3Department of Physiology, University of Toronto, Toronto, ON, Canada, 4Biology, University of Toronto Mississauga, Toronto, ON, Canada, 5Department of Cell & Systems Biology, University of Toronto, Toronto, ON, Canada, 6Edward S. Rogers Sr. Department of Electrical & Computer Engineering, University of Toronto, Toronto, ON, Canada
Synopsis
Tissue engineering with transplanted cells has the potential to repair and regenerate almost every tissue and organ of the body. One major obstacle of cell therapies is the inability to longitudinally assess injected cells. Non-invasive imaging with contrast-enhanced MRI is highly suited for this task but is limited with current methods. In this study, we report a novel method for producing bright endogenous cellular contrast through a genetic MRI reporter that results in the formation of in situ ferritin-manganese nanoparticles. The signal produced by these cells is significantly higher than traditional iron labelled ferritin-overexpressing cells and manganese-permeable cell lines.
Introduction
Cell therapy attempts to regenerate damaged tissue with exogenous cells have shown promise but suffer from the inability to directly probe cell fate and quantify injection success. MRI-based cell tracking approaches have been developed to address this issue; however, a vast majority utilize exogenous contrast agents to label cells prior to injection, which provides limited monitoring duration as the cell divides and signal is diluted. In this study, we report a novel genetic based method utilizing ferritin and manganese to produce sustained cellular contrast, with significant enhancement over current genetic-based MR methods.Methods
Human embryonic kidney cells (HEK293T) were transfected with CRISPR/Cas9 plasmids containing genes for divalent metal transporter 1 (DMT1) or ferritin. Transfected cells were selected by fluorescence sorting and singly seeded to produce monoclonal lines. Gene overexpression was determined by both western blotting and qPCR. In-vitro MRI was conducted with a 32-channel head coil on a 3.0T scanner. Cells were labelled with 0.05-1mM of manganese chloride (MnCl2) or ferric ammonium citrate (FAC) for 1, 24, 48 and 72 hours. T1-mapping was performed using an inversion recovery TSE sequence with a series of inversion times1. Transmission electron microscopy (TEM) was used to assess the formation of ferritin nanoparticles. Cells were incubated with 0.1mM MnCl2 for 24 hours before imaging or purification by immunoprecipitation with ferritin antibodies. Animal studies were performed on nine female NOD/SCID mice weighing 20-25g. Ferritin or DMT1-overexpressing cells (3x106) were injected intramuscularly into the gastrocnemius. Wild-type cells were injected in the contralateral leg. Before imaging, animals were injected subcutaneously with 0.4mmol/kg MnCl2 or orally supplemented with iron at 1–2.5mmol/kg. Animals were imaged using an 8-channel wrist coil on a 3.0T scanner. Coronal 2D fat-suppressed T1-weighted spin-echo images were acquired with in-plane resolution of 1mm x 0.3mm x 0.3mm, TR=507ms, TE=17.6ms, NSA=2. Quantitative T1-maps were acquired using non-selective T1 fast field echo using a variable flip-angle method2. Quantitative T2-maps were acquired using MS TSE with TR=2000 and 6 echoes. Quantitative T2*-maps were acquired using MS FFE with TR=100ms, FA=20, and 32 echoes. Animals were imaged longitudinally over the course of a week and then sacrificed for gross dissection and histological staining. Differences in relaxation times were determined using ANOVA. Post-hoc analysis was based on the Tukey-Kramer test. Significance was reported at a p-value of 5%.Results
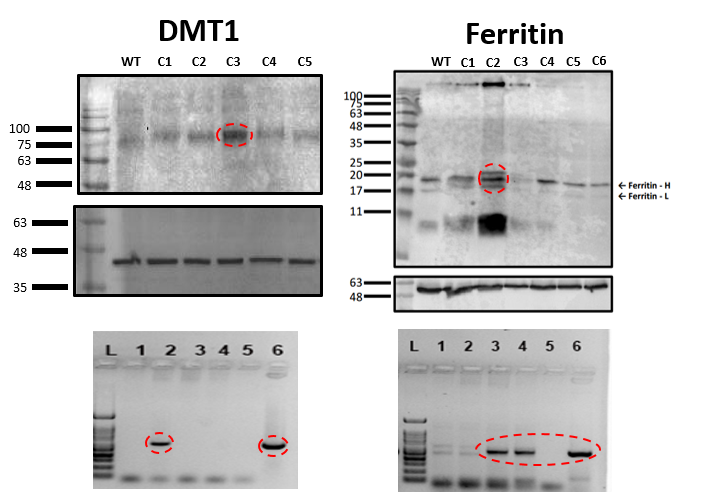
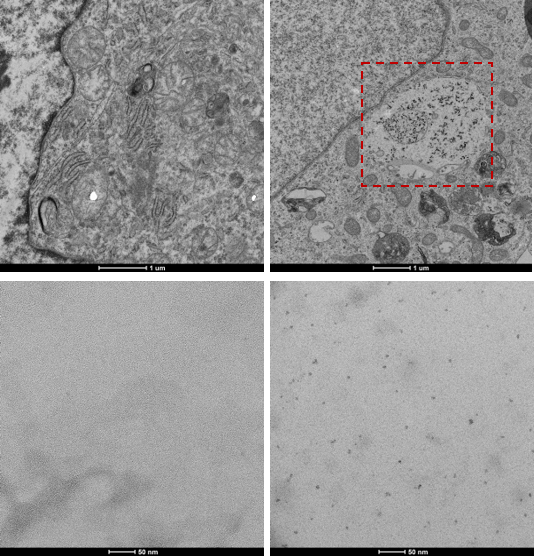
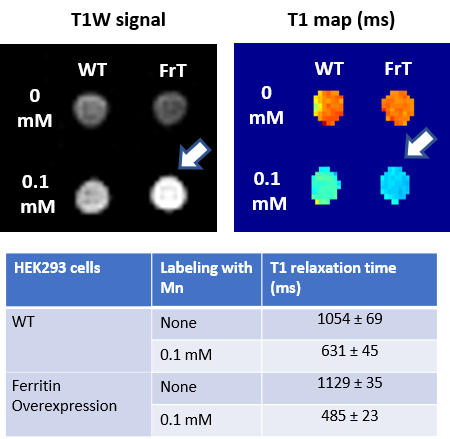
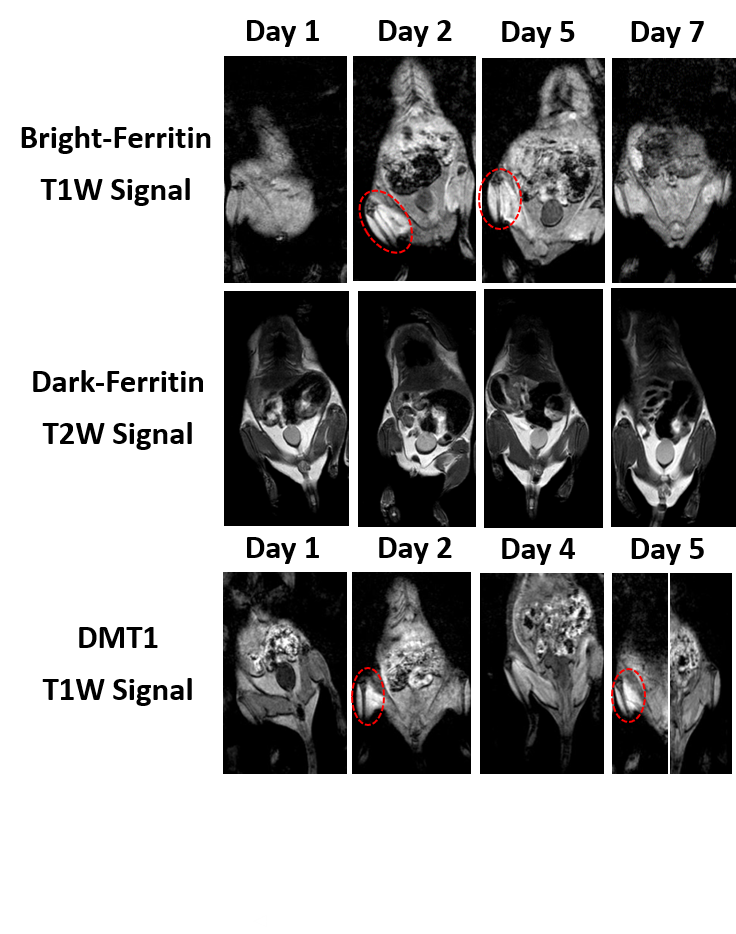
Ferritin and DMT1 genes were successfully inserted into the genome of HEK293T cells and clones were selected by protein overexpression (Fig.1). TEM of ferritin-overexpressing cells incubated with manganese produce visibly distinguishable amounts of nanoparticles that are electron dense (Fig.2) while wild-type cells do not. Furthermore, cell lysates immunoprecipitated with ferritin antibody contain intact ferritin nanoparticles with dense metallic cores measuring 5.4+0.3nm (Fig.2). Ferritin-overexpressing cells incubated with an optimized dose of 0.1mM MnCl2 for 24 hours exhibit significant T1 reductions of 24% (Fig.3) from wild-type cells and bright contrast on a T1-weighted image. In the presence of iron (FAC), (optimized dose 0.5mM, 48 hours), these cells exhibit a significant T2 reduction of 15%, while DMT1-overexpressing cells incubated with 0.1mM MnCl2 for 24 hours exhibit a T1 reduction of 41%. In-vivo cell tracking in mice was performed with wild-type, ferritin and DMT1-overexpressing cells injected in contralateral leg muscles for the duration of a week (Fig.4). Subcutaneous injections of MnCl2 24 hours prior to imaging exhibit cell site specific T1 reductions from 1050±57ms to 439±31ms with ferritin-overexpressing cells and 527±53ms with DMT1-overexpressing cells. This large change in T1 resulted in significant T1-weighted signal enhancement between contralateral legs injected with wild-type cells (Fig.4). Legs injected with ferritin-overexpressing cells maintained significant signal enhancement for up to 5 days, while legs injected with DMT1-overexpressing cells were no longer enhanced by day 4 and required an additional dose of manganese to produce signal again (Fig. 4). Animals injected with ferritin-overexpressing cells with or without oral iron supplementation did not produce significant changes in T2 or T2* throughout the duration of the week. The lack of difference in T2 and T2* resulted in indistinguishable T2-weighted signal between the contralateral legs (Fig.4). No mice suffered toxic or lethal effects from the injected cells or contrast dosing.Discussion
Ferritin-overexpressing cells in the presence of manganese produce bright and sustained contrast compared to wild-type cells, DMT1-overexpressing cells, and ferritin-iron cells in-vivo. These bright-ferritin cells contain nanoparticles with an electron dense metallic core and dimensions in-line with endogenous ferritin nanocages (diameter 5.5nm)3. These metallic nanoparticles are suspected to be composed of manganese due to the bright cellular contrast they produce. The build-up of Mn-Ferritin nanoparticles is suspected to be the cause for the sustained T1-weighted signal observed in-vivo in comparison to DMT1-overexpressing cells. In-vitro, DMT1-overpressing cells produce greater contrast due to manganese saturation. In-vivo however, as manganese is excreted from the body, equilibrium pressures reverse, and free manganese may leave the cell via the DMT1 channel. The incorporation of manganese into ferritin nanoparticles would retard this movement and result in sustained signal over time. These observations have been further validated in additional cell lines in our laboratory and provide great promise as a sensitive cell tracking tool.Conclusion
We have discovered, optimized and present a promising new MR genetic reporter complex for sensitive MRI tracking of cells.Acknowledgements
Funding from the University of Toronto’s Medicine by Design initiative which receives funding from the Canada First Research Excellence Fund, the Canada Foundation for Innovation Fund, Ontario Research Fund, Natural Sciences and Engineering Research Council Discovery Grant, UTM start-up fund, and Ontario Graduate Scholarships.References
1. Venter, A. et al. A manganese porphyrin-based T1 contrast agent for cellular MR imaging of human embryonic stem cells. Sci. Rep. 8, 12129 (2018).
2. Cheng, H.-L. M. & Wright, G. A. Rapid High-Resolution T1 Mapping by Variable Flip Angles: Accurate and Precise Measurements in the Presence of Radiofrequency Field Inhomogeneity. Magn. Reson. Med. 55, 566–576 (2006).
3.Watabe, T. & Hoshino, T. Observation of Individual Ferritin Particles by Means of Scanning Electron Microscope. J. Electron Microsc. (Tokyo). 25, 31–33 (1976).
Figures