4667
Functional connectivity MRI in head-fixed awake mice differentiates acute and chronic neuroinflammation1Biomedical Research Imaging Center and Department of Neurology, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States, 2Biomedical Engineering, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
Synopsis
Differentiating between acute and chronic neuroinflammation could improve clinical diagnoses of Alzheimer's disease, multiple sclerosis, and Parkinson's disease as well as disorders with no established clinical diagnostic methods such as Chronic Traumatic Encephalopathy and Chronic Fatigue Syndrom. This study implements mouse models of acute and chronic neuroinflammation to examine how neuroinflammation alter fcMRI in awake mice.
Introduction
Acute neuroinflammation is a protective response that diverts the brain’s energy to isolate, repair or destroy injured and infected brain cells [1] while inducing sickness behavior to conserve energy [2]. Chronic neuroinflammation is a pathogenic response involved in collateral neurodegeneration [3] and ‘depression-like’ behavior [2] associated with Alzheimer's disease, multiple sclerosis and Parkinson's disease [4]. The expression profile of immediate early genes (IEG) suggests that acute neuroinflammation suppresses neuronal activity except in Solitary Nucleus Tract (SNT), locus coeruleus (LC), the nucleus acumens (NAc), paraventricular thalamus (PVT), amygdala and lateral hypothalamus (LH) [5-7]. Though chronic neuroinflammation has been shown to alter the hedonic network [8], other regions remain to be explored and validated using animal models. We performed fcMRI on mice exposed to low or high dose of the endotoxin lipopolysaccharide (LPS) to induce acute or chronic neuroinflammation, respectively [9]. This study asked whether fcMRI is sensitive enough to differentiate brain network changes associated with acute and chronic neuroinflammation. We had two major innovations in this study: (1) a novel head-fixed protocol for fcMRI in fully awake mice with integrated physiological monitoring; and (2) a novel pCREB histological protocol to verify neuroinflammation-mediated fcMRI changes were attributed to altered neuronal activation rather than vascular effects.
Methods
Adult C57BL6/J mice were exposed to saline vehicle (n=12), 3 × 106 EU/kg of LPS ( E. coli strain O111:B4, Sigma-Aldrich L3012; n=12), or 15 × 106 EU/kg of LPS (n=12). Changes in ambulation and body temperature were assessed to verify sickness behavior. A subset of mice were euthanized without fixation, their blood and brains extract at 3 hours and 7 days (n=3/exposure/endpoint) following exposures to confirm neuroinflammation. Serum was extracted from the blood for protein quantification. The left hemisphere was homogenized in 100 mg tissue/ml cold lysis buffer (20 mM Tris, 0.25 M sucrose, 2 mM EDTA, 10 mM EGTA, 1% Triton X-100) with 1 Complete Mini protease inhibitor cocktail tablet (Roche Diagnostics, Indianapolis, IN) / 10 ml of lysis buffer and spun at 100,000 × g for 40 min to extract brain protein for quantification. TNFα was measured by ELISA (R&D Systems, Minneapolis, MN). Right hemispheres were post-fixed in 4% PFA for 48 h, dehydrated in 30% sucrose for 48 h, and frozen in OCT compound prior to being cut into 35 μm sagittal sections for immunofluorescent histology. Neuroinflammation was confirmed using Iba-1 antibody to assess microglia morphology and neuronal activity was confirmed using pCREB antibody. Prior to performing fcMRI, a plastic head mount was superficially implanted on the mice and they were habituated in the awake holder by performing scans for 8 consecutive days until their body temperatures stabilized. A Bruker 9.4-Tesla/30-cm scanner with a homemade mouse transceiver surface coil was used for acquisition. Single-shot gradient echo EPIs were acquired in two, 10 minute sessions with bandwidth = 250 kHz, TR/TE = 3000/10 ms, matrix = 64x64, FOV = 1.92 cm2, slice number = 26 and slice thickness = 0.3 mm. BOLD acquisitions were slice-timing and motion corrected, aligned, spatial smoothed (FWHM = 0.6 mm), bandpass filtered (0.01-0.1 Hz) and normalized using FSL and warped with ANTs to a custom C57BL/6J probabilistic EPI atlas. Correlation maps were generated using Pearson correlations undergoing a Fisher's r-to-Z transformation through JMP (v.13, SAS) and visualized on a connectivity matrix.
Results and Discussion
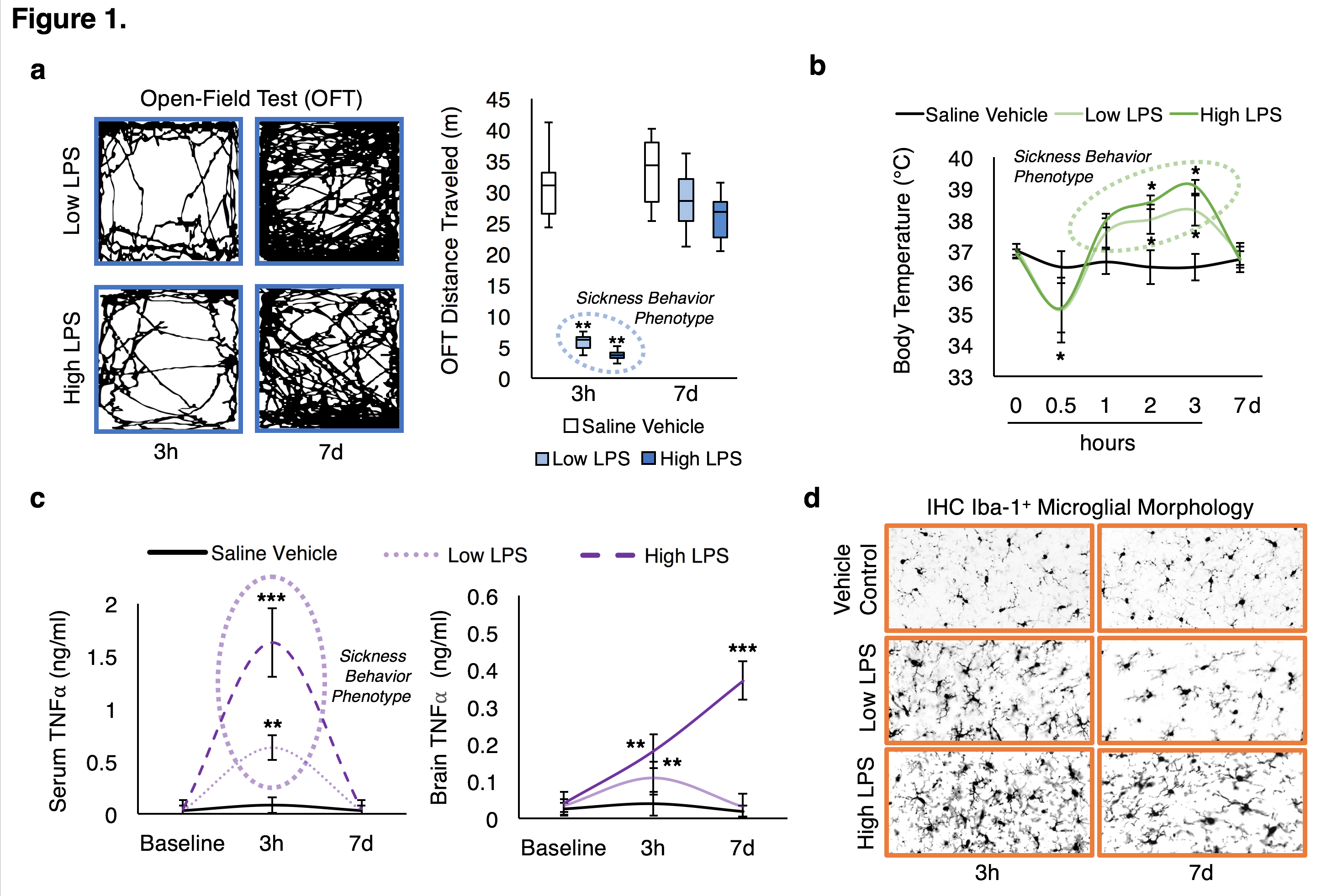
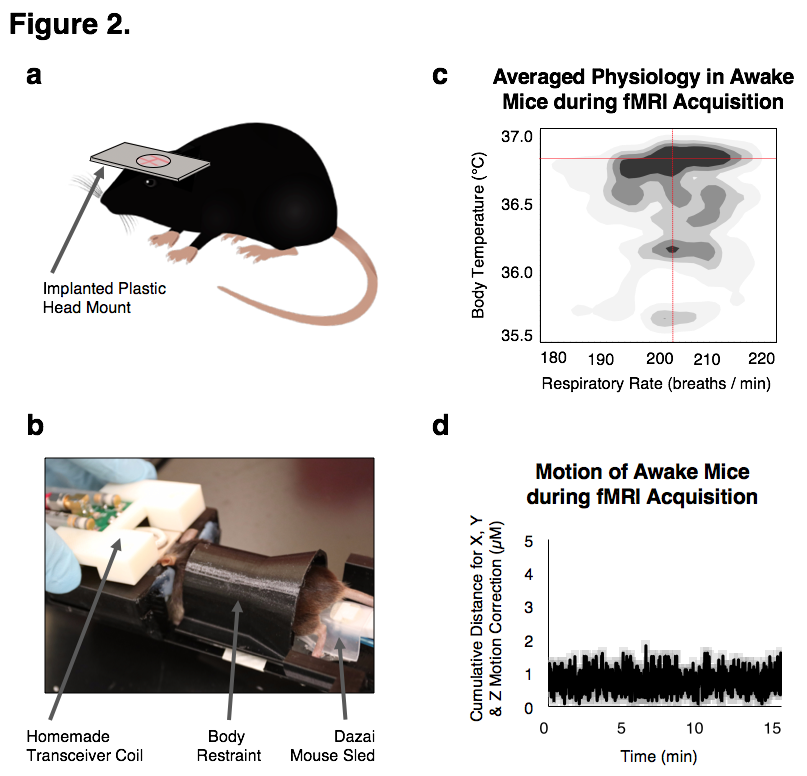
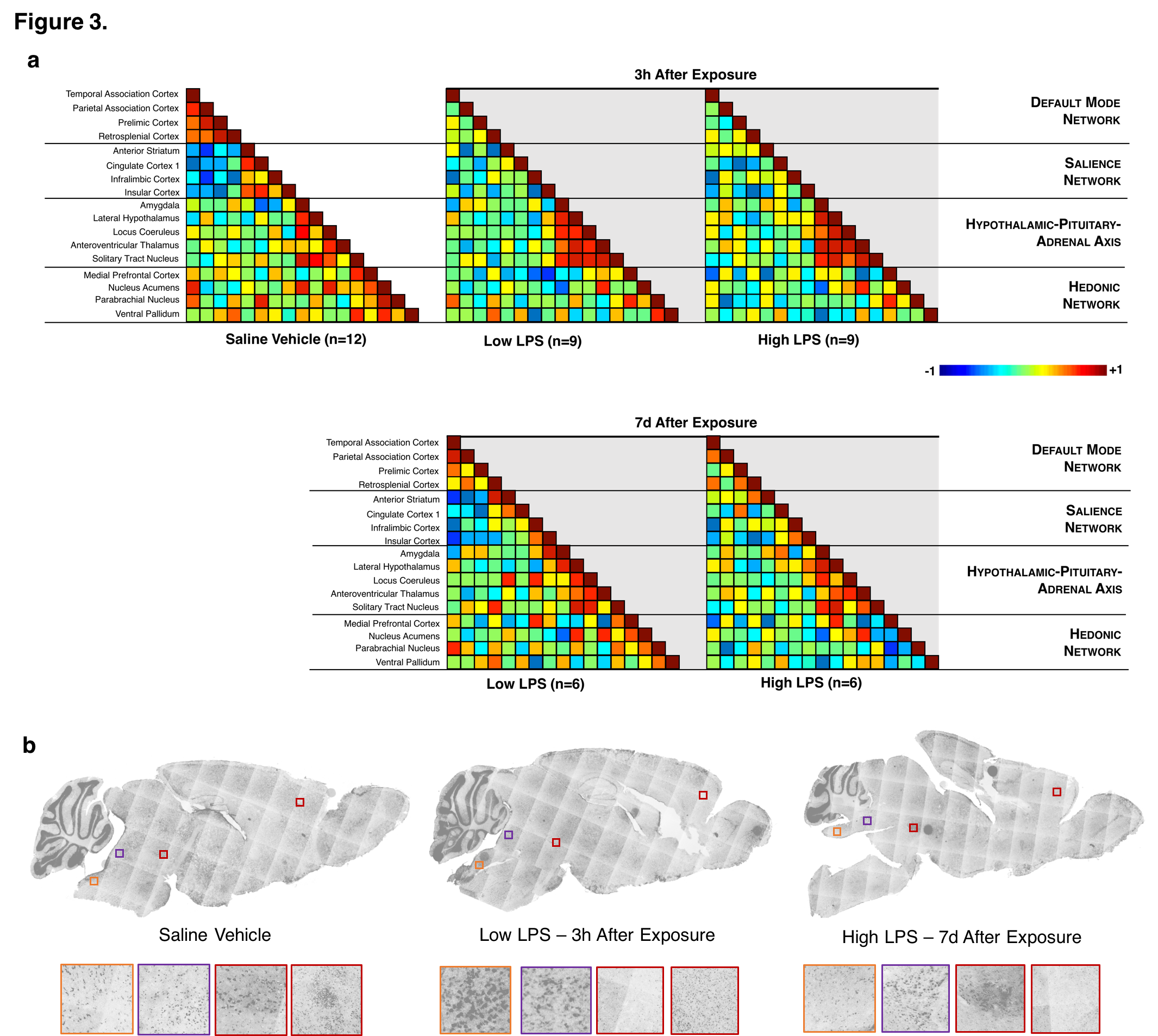
We successfully generated mouse models of acute and chronic neuroinflammation by altering the exposure dose of LPS. Mice exposed to both doses induced sickness behavior (Fig 1A, B) and had significantly elevated levels of TNFα in their blood (Fig 1C). Though levels of TNFα in the brain were significantly elevated for both doses at 3h following exposure, only high LPS dose mice showed elevated TNFα (Fig 1C) and microglial morphologies indicative of being immunologically activated (Fig 1D) at 7d following exposure. Awake fcMRI was performed using a custom built head-fixed holder and head mount (Fig 2A, B) allowing for stable physiology (Fig 2C) and almost no head movements (Fig 2D). Connectivity matrices and pCREB histology indicate that both LPS doses globally suppress neuronal activation at 3h following exposure, limiting neuronal activity to the hypothalamic-pituitary-adrenal axis (Fig 3A, B). By 7d following exposure, low LPS dosed mice regain connectivity patterns and pCREB+ neuronal activation similar to the saline vehicle while high LPS dosed mice maintain global suppression of neuronal activity and had an inverse correlation among the hedonic networks--indicating a putative state of anhedonia. Our findings suggest that fcMRI can be used to differentiate acute from chronic neuroinflammation which could otherwise go clinically undetected due to their similar symptoms and functional connectivity patterns during onset.
Acknowledgements
We thank the members of the Shih lab for their valuable discussions regarding the studies described in this abstract. Our team is supported by NIMH R01MH111429, R41MH113252, R21 MH106939, NINDS R01NS091236, NIAAA U01AA020023, R01AA025582, NICHD U54HD079124, American Heart Association 15SDG23260025, and Brain & Behavior Research Foundation. E.A.O. was directly supported by NHLBI T32HL069768.References
[1] Ousman, S.S. and P. Kubes, Immune surveillance in the central nervous system. Nat Neurosci, 2012. 15(8): p. 1096-101.
[2] Dantzer, R., et al., From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci, 2008. 9(1): p. 46-56.
[3] Block, M.L., L. Zecca, and J.S. Hong, Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci, 2007. 8(1): p. 57-69.
[4] Ransohoff, R.M., How neuroinflammation contributes to neurodegeneration. Science, 2016. 353(6301): p. 777-83.
[5] Park, S.M., R.P. Gaykema, and L.E. Goehler, How does immune challenge inhibit ingestion of palatable food? Evidence that systemic lipopolysaccharide treatment modulates key nodal points of feeding neurocircuitry. Brain Behav Immun, 2008. 22(8): p. 1160-72.
[6] Dallaporta, M., et al., c-Fos immunoreactivity induced by intraperitoneal LPS administration is reduced in the brain of mice lacking the microsomal prostaglandin E synthase-1 (mPGES-1). Brain Behav Immun, 2007. 21(8): p. 1109-21.
[7] Conde, G.L., et al., Central LPS-induced c-fos expression in the PVN and the A1/A2 brainstem noradrenergic cell groups is altered by adrenalectomy. Neuroendocrinology, 1999. 70(3): p. 175-85.
[8] Felger, J.C., et al., Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol Psychiatry, 2016. 21(10): p. 1358-65.
[9] Qin, L., et al., Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia, 2007. 55(5): p. 453-62.
[10] Simon, D.W., et al., The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol, 2017. 13(3): p. 171-191.
[11] Papadopoulos, V. and L. Lecanu, Translocator protein (18 kDa) TSPO: an emerging therapeutic target in neurotrauma. Exp Neurol, 2009. 219(1): p. 53-7.
Figures