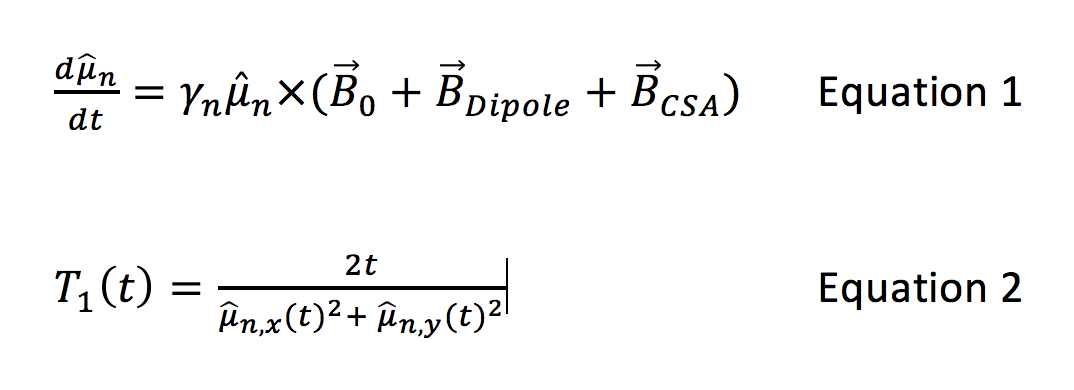4022
Use of Ab-Initio Molecular Dynamics Simulations to Estimate Hyperpolarized 13C Agent Spin-Lattice Relaxation Times1Radiology, University of Pennsylvania, Philadelphia, PA, United States
Synopsis
We demonstrated the feasibility of using an ab-initio molecular dynamics simulation to estimate the T1 relaxation time constant of hyperpolarized 13C substrates. The utility of such predictive tools would provide further insight into specific relaxation mechanisms involved in various substrates, allowing us to evaluate candidate strategies to extend their T1 relaxation time constants. Such screening of candidate hyperpolarized agents may be used to rapidly assess polarizability of agents specific to various molecular pathways.
Introduction
Injected hyperpolarized agents show great promise for highlighting disorders in which metabolic activity is locally altered. However, agent spin-lattice relaxation limits the practicality of this technique in some otherwise useful imaging applications. Although overall trends with molecular size and nuclear environment are well known, it is generally difficult to predict longitudinal relaxation time with any accuracy and thereby rationally select agents with a reasonable likelihood of success. A reliable in silico predictive tool would allow rapid screening of candidate agent/pathway combinations, would clarify the relative importance of relaxation mechanisms, and would allow evaluation of candidate strategies for T1 extension (e.g., deuteration, modification of solvent, or use of long-lived, multinuclear states).Materials and Methods
Because of their importance or potential in the hyperpolarized imaging community, predictions were made for four molecules; the pyruvate and acetate ions, glucose (neutral β-D-glucopyranose) and cysteine (neutral zwitterionic). Molecular geometries were designed graphically using Avocadro 1.2.0 and allowed to relax to lowest energy geometry using a steepest-descent electrostatic algorithm. Molecular dynamics simulations were performed using Gromacs 2016.3. Molecules in isolation were solvated with TIP4P water molecules (H2O or D2O) in a periodic boundary condition cube measuring 2.3nm on a side. One Na+ was added as necessary to ensure overall neutrality. An initial system-wide energy minimization was followed by 1ns of thermal equilibration starting from random velocities consistent with T=298.15K. Finally, a 10ns molecular dynamics simulation was performed at constant temperature and 1 bar pressure using the Nose-Hoover thermostat and Parrinello-Rahman pressure coupling. Force integration and coordinate/velocity information was updated every femtosecond, with coordinate output every picosecond to form a single system trajectory of 10000 timepoints. Trajectories were post-processed to eliminate nonphysical interaction fluctuations due to jumps across the box boundary. After the trajectory of each nucleus was calculated as above, the chemical shift tensor was separately calculated at each timepoint for the nucleus of interest using Gaussian 09 with B3/LYP unrestricted density functional theory and the 6-31 basis set. To limit computational time, each Gaussian calculation was restricted to the target molecule and waters within 0.5nm. Three candidate interactions were considered in calculating the relaxation. Target nuclei were initially placed in the fully-aligned state. Dipole-dipole interactions were included by assigning other nuclei random classical orientations and recalculating temporally fluctuating dipolar fields at each timestep. CSA fields were included using the chemical shift tensor appropriate to each timestep’s electronic configuration and molecular orientation. The resulting fields were summed and used to evolve the target nucleus orientation as Equation 1 (see last figure).
Although this calculation is classical, any deviation from the fully quantum calculation involves multi-nuclear coherences that we expect to be entirely negligible. For times long compared to the relevant correlation time, T1 can then be estimated using the change in as <μn,z> as Equation 2 (see last figure).
Contribution of intramolecular scalar interactions were estimated using Gaussian-derived couplings; the overall small size of the interactions and short correlation time of their fluctuations were such that their effect was deemed negligible in all cases studied. Experimental T1 values were measured using sealed, 80mM, 3x freeze-thaw degassed H2O/D2O solutions with approximately 500mM of EDTA to chelate metal ions.
Results
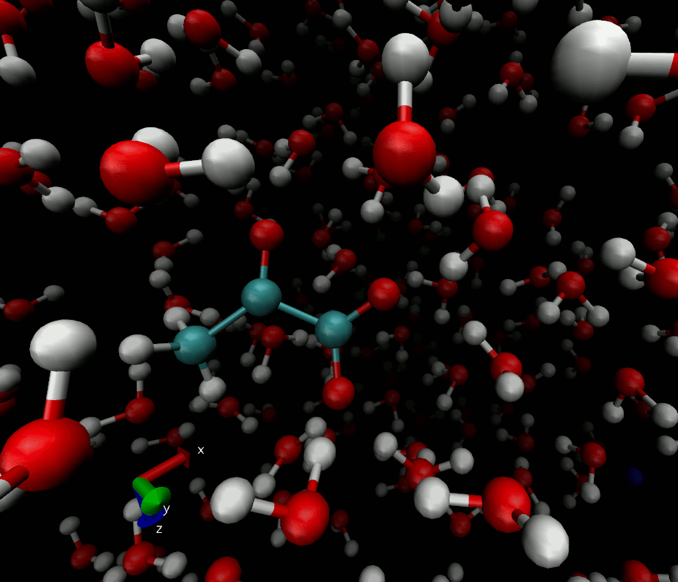
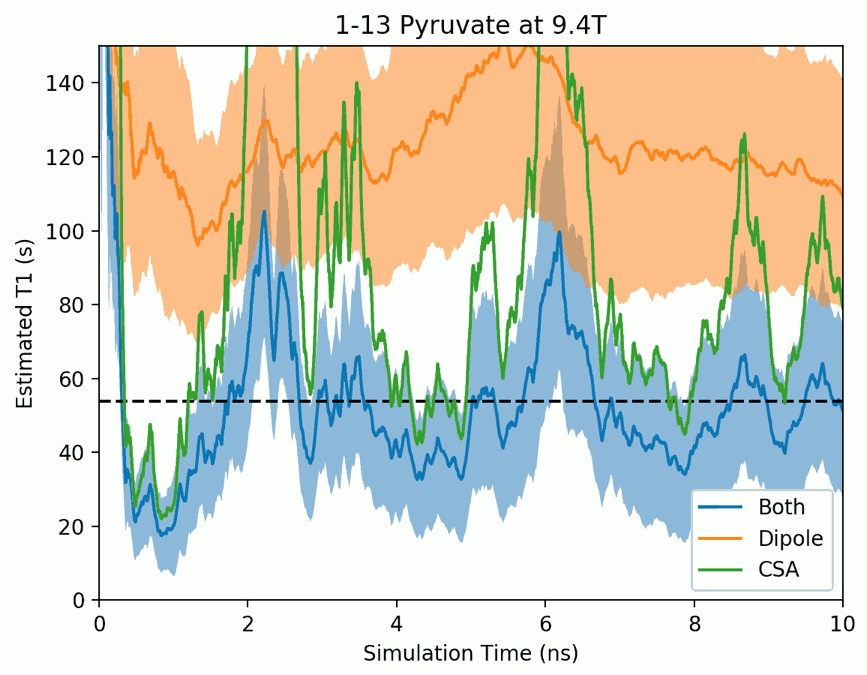
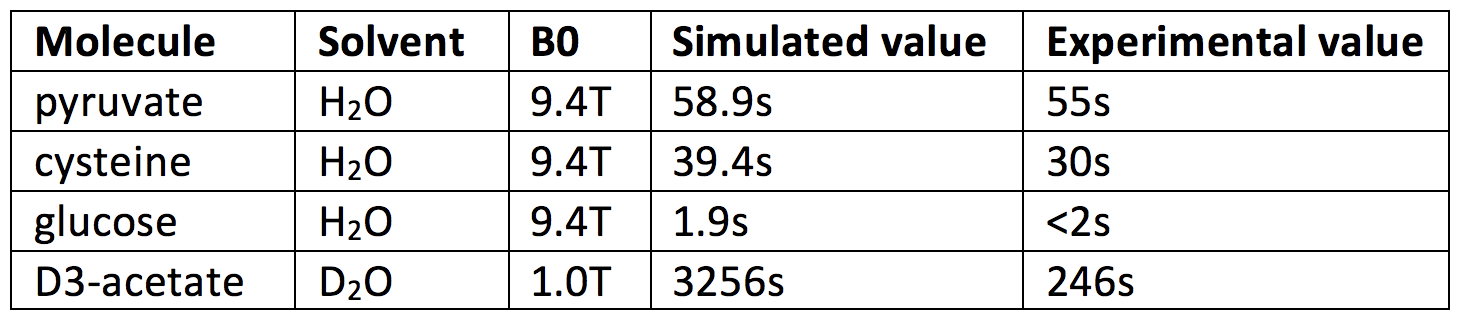
Figure 1 depicts water, target ion and counterion positions for one timestep of the pyruvate T1 simulation. The simulated region contains approximately 400 water molecules which we find to be sufficient to adequately capture the altered molecular motion surrounding the molecules of interest studied. Figure 2 shows the evolution of the simulated T1 during a single pyruvate trajectory; note that because of significant fluctuation along a single trajectory, reported results represent the average of at least 1000 different trajectories started with random initial positions and nuclear orientations. Table 1 summarizes the calculated and measured T1 values at the experimentally available fields of 1.0 and 9.4T.Discussions and Conclusions
Of the moieties addressed so far, agreement between calculated and measured T1 values is generally excellent when CSA and dipolar relaxation mechanisms dominate (Table 1, rows 1-3). This suggests that the molecular dynamics and resulting correlation times reproduce those of the solvated molecule with acceptable accuracy. When dipolar interactions are minimized through deuteration and CSA is minimized through use of low field (Table 1, row 4), the simulation predicts very slow T1 relaxation which is not observed. Effect size estimations suggest that neither scalar coupling nor modulation of the CSA tensor during protonation/deprotonation can be responsible for this discrepancy. Future work will focus on identifying remaining mechanism(s) and applying the technique to other molecules and nuclear states relevant to hyperpolarized imaging.Acknowledgements
No acknowledgement found.References
No reference found.Figures