3657
Clinico-genetic-anatomical comparisons of paroxysmal kinesigenic dyskinesia with and without PRRT2 mutations1Department of Radiology, Huaxi MR Research Center, Chengdu, China
Synopsis
Paroxysmal kinesigenic dyskinesia (PKD) is a rare movement disorder characterized by sudden, brief attacks of involuntary movements. This study aims to detect the topological organization of white matter structural connectivity in PKD with and without PRRT2 mutations using graph theoretical approaches. Compared with non-PRRT2 mutation carriers, PRRT2 mutations carriers are significantly associated with a younger age of onset, a complicated form of PKD, combined phenotypes of dystonia and chorea, and a tendency for a family history of PKD in our population, as well as showed topological trends for randomization.
Introduction
Paroxysmal kinesigenic dyskinesia (PKD) is a rare movement disorder characterized by sudden, brief attacks of involuntary movements that are precipitated by sudden voluntary movement. Though there are no apparent morphological changes, the dysfunction of the basal ganglia-thalamocortical pathway are proposed as the neuroanatomical basis of the disease1. Recently PRRT2 (proline-rich transmembrane protein 2) gene has been identified as a causative gene of PKD , there are still a quite proportion of PKD patients identified without the mutation of PRRT2, and the pathogenic mechanism of the mutation remains largely unknown. PKD patients with PRRT2 mutations are usually associated with an earlier age at onset, longer duration of attacks, a complicated form of PKD, combined phenotypes of dystonia and chorea, and a tendency for a family history of PKD2, however, the effect of PRRT2 mutation on anatomical networks in PKD patients remains largely unknown. This study aims to detect the topological organization of white matter structural connectivity in PKD with and without PRRT2 mutations using graph theoretical approaches.Methods
We recruited 20 idiopathic PKD patients with PRRT2 mutations, and 26 without PRRT2 mutations. Detailed clinical information on demographics, nature of attacks, family history and the presence of associated conditions were obtained by in-person interviews using a standardized questionnaire. We extracted DNA from blood samples of the study subjects. MR imaging data were acquired with a 3.0-T MR imaging system by using a single-shot spin-echo echoplanar image sequence that included one high-resolution T1 image and one DT imaging data image. All the image preprocessing and analyses were implemented by using PANDA (a pipeline tool for diffusion MR imaging). The structural connectome was constructed by using DT imaging tractography and thresholding the mean fractional anisotropy of 90 brain regions to yield 90 × 90 partial correlation matrixes. Graph theory analysis was used to examine the group-specific topologic properties by using the GRETNA toolbox investigating the topologic properties of brain networks at both the global and nodal level, and nonparametric permutation tests were used for group comparisons of topologic metrics.Results
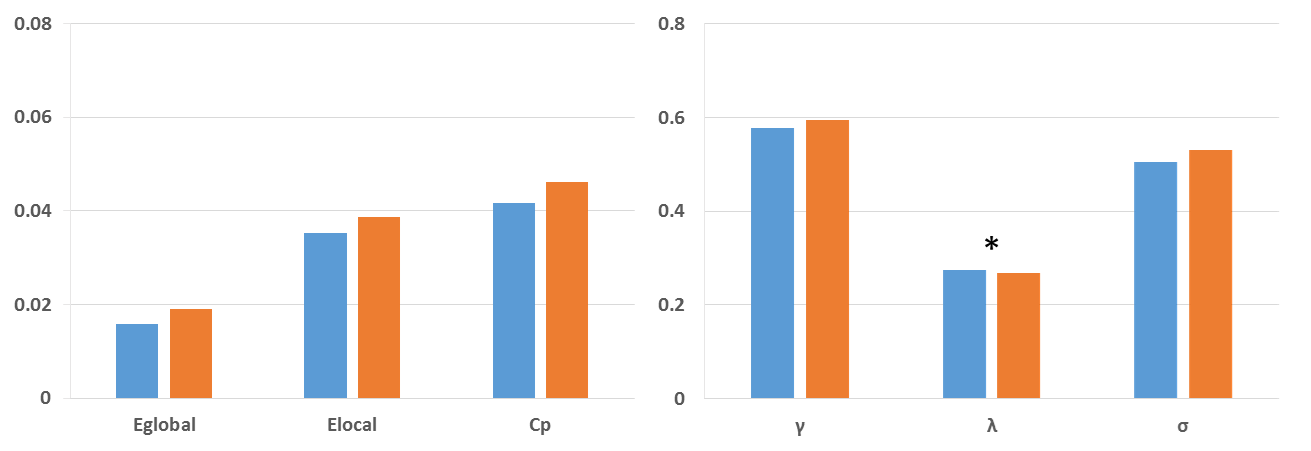
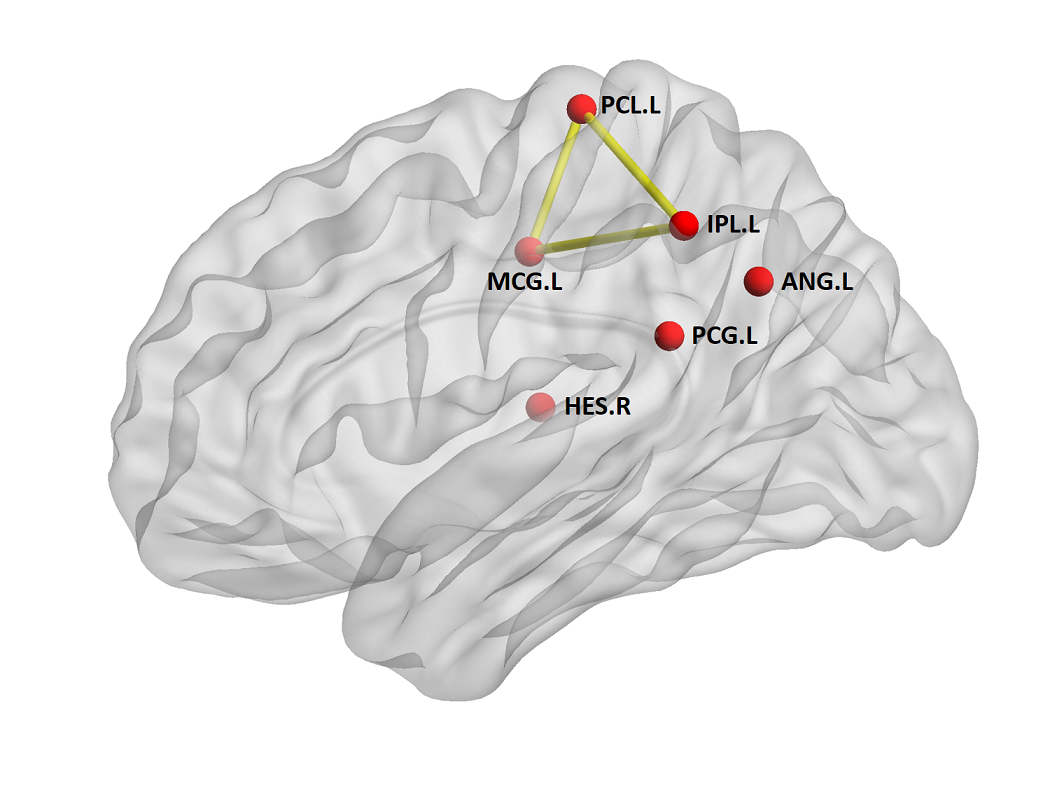
Compared with non-PRRT2 mutation carriers (mean age at initial onset was 12.5 ± 2.8 years), the PRRT2 mutation carriers were younger at onset (mean age at initial onset was 10.7 ± 5.6 years); and present with complicated PKD, combined phenotypes of dystonia and chorea. Moreover, 15 among 20 PRRT2 mutation carriers reported a positive identified or suspected family history, while only one non-PRRT2 mutation carrier reported his mother has a family history of seizures. Both groups exhibited small-world topology. However, PRRT2 mutation carriers showed topological trends for randomization, demonstrated as lower normalized characteristic path length λ (P = 0.017) compared with controls. Furthermore, the patients demonstrated significantly decreased nodal centralities in the left paracentral lobule, left inferior parietal lobule, left angular gyrus, right Heschl’s gyrus and left middle and post cingulum. The PKD-related alterations in anatomic connections involving in the paracentral- inferior pariental- middle cingulum circuit.Discussion
Consistent with previous studies, PRRT2 mutations carriers are significantly associated with a younger age of onset, a complicated form of PKD, combined phenotypes of dystonia and chorea, and a tendency for a family history of PKD in our population. Meanwhile, the PRRT2 mutation carriers showed significant decrease in the normalized characteristic path length, suggesting a shift toward random network. In addition to the global topologies, one of the decreased nodal activities we observed was the left paracentral lobule, which is very important in the movement disorder since it controls motor and sensory innervations of the contralateral lower extremity. As dystonia is commonly viewed as a sequence of motor dysfunction, our results provided an anatomic basis for the pathophysiological mechanisms of PKD.Conclusion
In conclusion, from the clinico-genetic-anatomic aspects, our studies comparing PKD patients with and without the mutation of PRRT2 provide topologic insights into understanding the pathophysiological mechanisms of PKD. However, direct correlations and interactions of the clinico-genetic-anatomic factors should be investigated in future.Acknowledgements
No acknowledgement found.References
1, Long Z et al., Thalamocortical dysconnectivity in paroxysmal kinesigenic dyskinesia: Combining functional magnetic resonance imaging and diffusion tensor imaging. Mov Disord. 2017 Apr;32(4):592-600
2, Huang XJ et al., Paroxysmal kinesigenic dyskinesia: Clinical and genetic analyses of 110 patients. Neurology. 2015 Nov 3;85(18):1546-53
Figures