3650
The Protective Effects of Ultramicronized Palmitoylethanolamide on the glutamatergic system in a triple transgene mouse model of Alzheimer disease1Dept. of Cell Biology and neurosciences, Istituto Superiore di Sanita', Rome, Italy, 2Dept. of Physiology and Pharmacology “V. Erspamer”, SAPIENZA University of Rome, Rome, Italy, 3Dept. of Clinical and Experimental Medicine, University of Foggia, Foggia, Italy
Synopsis
We investigate the effects of a chronic treatment with an endogenous lipid mediator palmitoylethanolamide on the onset and progression of AD in 3xTg-AD mice. Behavioural tests showed improvements in learning and memory, and in both the depressive and anhedonia-like phenotype in PEA-treated 3×Tg-AD mice. MRI/MRS in vivo analysis, microdialysis and western blot, RT-PCR, and immunofluorescence show that PEA normalizes astrocytic function, rebalances glutamatergic transmission, and restrains neuroinflammation. The efficacy of PEA is particularly potent in younger mice, suggesting its potential as an early treatment.
Introduction
The endogenous lipid mediator palmitoylethanolamide (PEA, a naturally occurring amide of ethanolamine and palmitic acid) has demonstrated exceptional potential as a novel treatment for Alzheimer disease (AD). Its anti-inflammatory and neuroprotective properties, as well as its ability to preserve memory function in rodent models of AD are well documented.1-5 The effects of chronic PEA administration on the progression of AD and the optimal time to begin treatment are still unknown.Purpose
In the present work we investigate the effects of a chronic treatment with ultramicronized PEA (um-PEA), a formulation that overcomes bioavailability concerns owing to its lipid nature, on the onset and progression of AD in 3xTg-AD mice who develop amyloid plaques and neurofibrillary pathology in AD-relevant brain regions (hippocampus cortex) and mimic the disease progression in humans.6-8Methods
We studied young (6-month-old, n=11) and adult (12-month-old, n=16) 3×Tg-AD (harboring APPswe, PS1M146V, tauP301L transgenes) male mice and their sex- and age-matched wild-type littermates (Non-Tg) (C57BL6/129SvJ) (n=13 and n=14, for 6 and 12 months, respectively) after a 3 months continuous administration of um-PEA (10mg/kg/day, or vehicle) via a release pellet subcutaneously. At the end of the treatment, mice were tested with a range of cognitive and non-cognitive tasks.
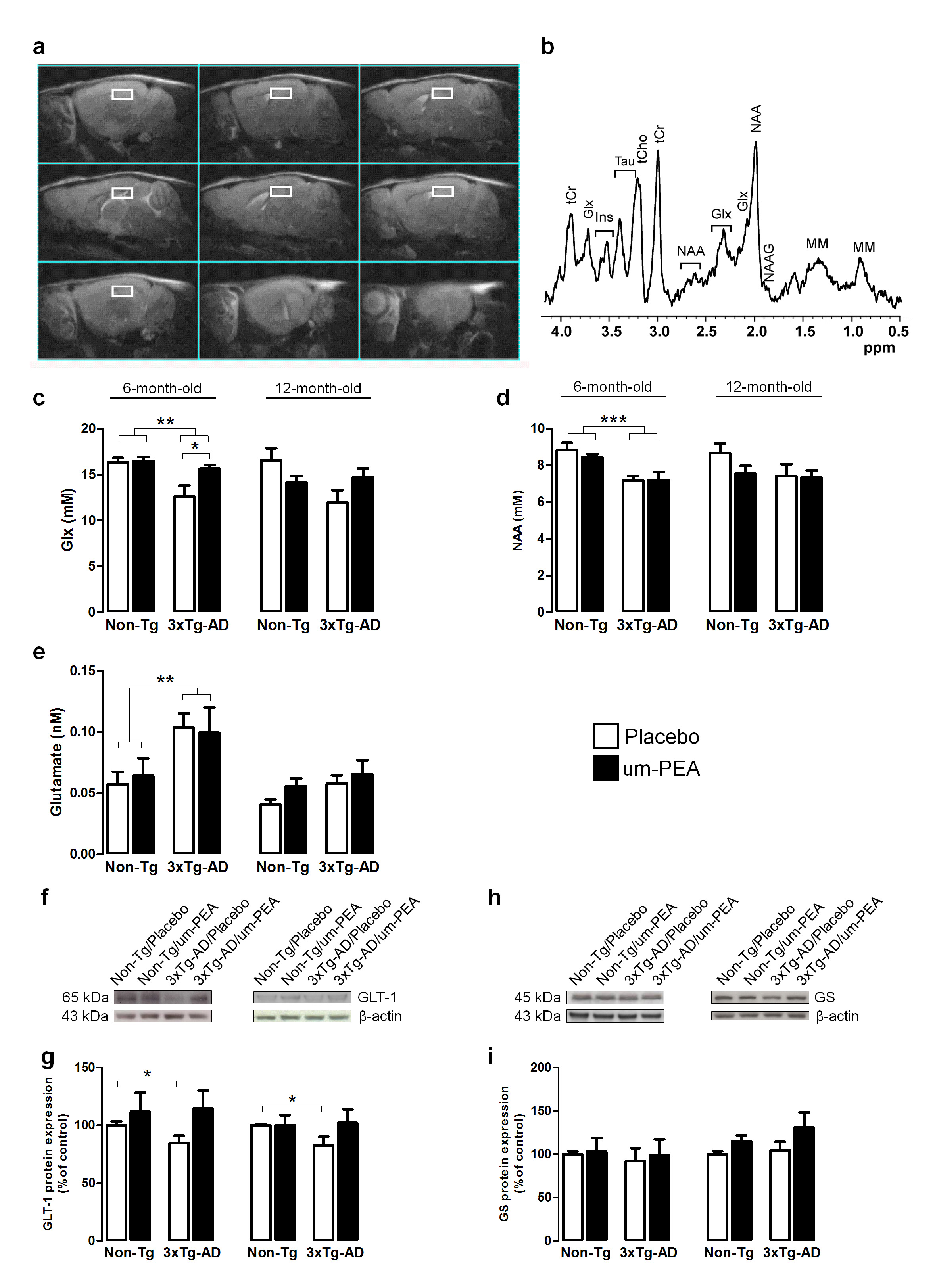
At the end of behavioural tests mice undergo MRI and MRS scanning to evaluate genotype- and treatment-induced differences in brain metabolism. MR examinations were performed on VARIAN Inova MRI/MRS system (4.7T) and a combination of volume and surface coil (RAPID Biomedical). Multislice fast spin-echo (TR/TEeff=3000/70ms, ns=2, slice thickness 1mm, matrix 128x256) sagittal images were acquired to localise the regions of interest. Single-voxel localised 1H MR spectra (PRESS,TR/TE=4000/23ms, ns=256) were collected from: prefrontal cortex (PFC), 10.8ml and hippocampus (Hip), 9.5ml. as shown in Fig. 1a,b. Spectra were analysed by using LCModel program and unsuppressed water signal as reference.9 Statistical analysis was performed by using ANOVA (2x2 genotype and treatment).
Neurochemical release was also assessed by microdialysis and potential neuropathological mechanisms were assessed post-mortem by RT-PCR, western blot, and immunofluorescence.
Results and discussion
Our data demonstrates that um-PEA improves learning and memory, and ameliorates both the depressive and anhedonia-like phenotype of 3×Tg-AD mice. Quantitative MRS analyses revealed that 6-month-old 3×Tg-AD mice have reduced hippocampal levels of both Glx (a combined measure of glutamate and glutamine both intra- and extra-cellular) and NAA compared to age-matched Non-Tg littermates (Fig.1c,d). Interestingly, um-PEA treatment significantly increases only Glx levels in the transgenic mice (Fig.1c) with the increase mainly due to Glu. Glx reflects the intracellular glutamate and glutamine pools. To gain a deeper insight into the functional state of glutamatergic transmission, we performed microdialysis experiments to evaluate extracellular glutamate levels. Results showed that basal extracellular glutamate levels in the hippocampus of 6-month-old 3×Tg-AD mice resulted significantly higher compared to age-matched Non-Tg animals (Fig.1e). Western blot analyses indicated that the higher extracellular glutamate levels in transgenic mice may be due to the reduced expression of GLT-1 (the glial transporter mainly responsible for glutamate re-uptake) while the expression of glutamine synthetase (an enzyme that plays an essential role in the metabolism of glutamate to form glutamine) resulted unaffected (Fig.1f-i). Even though not statistically significant, um-PEA treatment caused a trend toward an increase (+35%) of GLT-1 in 6-month-old 3×Tg-AD mice compared to placebo-treated 3×Tg-AD (Fig.1 g,h). In 12-month-old mice we did not record any genotype-related difference in Glx, NAA or extracellular glutamate levels even if the reduction of GLT-1 was still evident.
Collectively, these results indicate that 6-month-old transgenic animals exhibited alterations in the glutamatergic system and that um-PEA was able to increase Glx levels.
Conclusions
Our data demonstrates the first in vivo evidence that chronic treatment with um-PEA induces considerable improvements in cognitive and neural function during both the early pre-symptomatic and later symptomatic stages of AD in a triple transgenic mouse model of AD, a model which closely mimics the neuropathological alterations seen in human AD. Considering the safety and tolerability of PEA in humans, our findings offer new opportunity in AD treatment.Acknowledgements
This work was supported by the Italian Ministry of Instruction, University and Research grants (MIUR; PON01-02512 and PRIN2009)References
1. Scuderi C, et al. J Cell Mol Med 2011; 15: 2664-2674.
2. Scuderi C, et al. Cell Death Dis 2014; 5: e1419.
3. Scuderi C, et al. J Neuroinflammation 2012; 9: 49.
4. D'Agostino G, et al. Neuropsychopharmacol 2012; 37(7): 1784-1792.
5. Tomasini MC, et al J Alzheimers Dis 2015; 46(2): 407-421.
6. Oddo S, Caccamo A, Kitazawa M, et al. Neurobiol Aging 2003; 24, 1063–1070.
7. Oddo S, Caccamo A, Shepherd JD, et al. Neuron 2003b; 39, 409–421.
8. Billings LM, Oddo S, Green KN, et al. Neuron 2005; 45, 675– 688.
9. De Filippis B, Fabbri A, Simone D, Canese R, et al. Neuropsychopharmacology. 2012;37(5):1152-63.
Figures