3639
Motor compensation in Parkinson’s disease (PD): a multimodal neuroimaging study1Neurology, Groupe Hospitalier Pitié Salpêtrière, Paris, France, 2Institut du Cerveau et de la Moelle Epinière, Paris, France, 3Neuroradiology, Groupe Hospitalier Pitié Salpêtrière, Paris, France, 4Nuclear Medicine, Groupe Hospitalier Pitié Salpêtrière, Paris, France, 5Centre d'Investigation de Recherche Clinique, Groupe Hospitalier Pitié Salpêtrière, Paris, France
Synopsis
Motor compensation mechanisms in PD rely on very distinct levels of evidence and have never been assessed altogether. They were tested among 68 early PD patients who underwent dopaminergic imaging and MRI (T1 neuromelanin, fMRI, DTI). We created an adjusted motor severity index (ratio between akinetic motor severity and neuromelanin substantia nigra alteration) and performed correlations between this index and the hypothesized compensation mechanisms. As a result, new dopaminergic synapses or more active dopaminergic synapses, reorganization of motor and cognitive subcortical loops and of the associative areas of the cerebellum are the main motor compensation mechanisms in early PD.
INTRODUCTION
In PD, substantia nigra pars compacta (SNpc) neurons degeneration is thought to be responsible for striatal dopaminergic denervation that unbalances motor-subcortical loops activity and in the end generates motor akinetic symptoms. Nonetheless, numerous discrepancies have been observed between the degree of nigro-striatal denervation and the severity of motor symptoms1,2. These discrepancies have led to the hypothesis of motor compensation in PD. Several biological mechanisms have been assumed to underlie this hypothesis: dopaminergic synapse metabolism, new dopaminergic synapses, intra motor-subcortical loop activity reorganization, inter motor and cognitive subcortical loops activity reorganization, cerebellum and pedunculopontine nucleus activity3,4. Nevertheless, these mechanisms rely on very distinct levels of evidence (animal model, simple hyperactivity in neuroimaging studies…) and have never been assessed altogether in vivo in PD patients. Therefore, we propose to assess these mechanisms using a multimodal neuroimaging cohort of patients with clinical early PD and prodromal PD (idiopathic Rapid eye movement Behavior Disorder – iRBD).METHODS
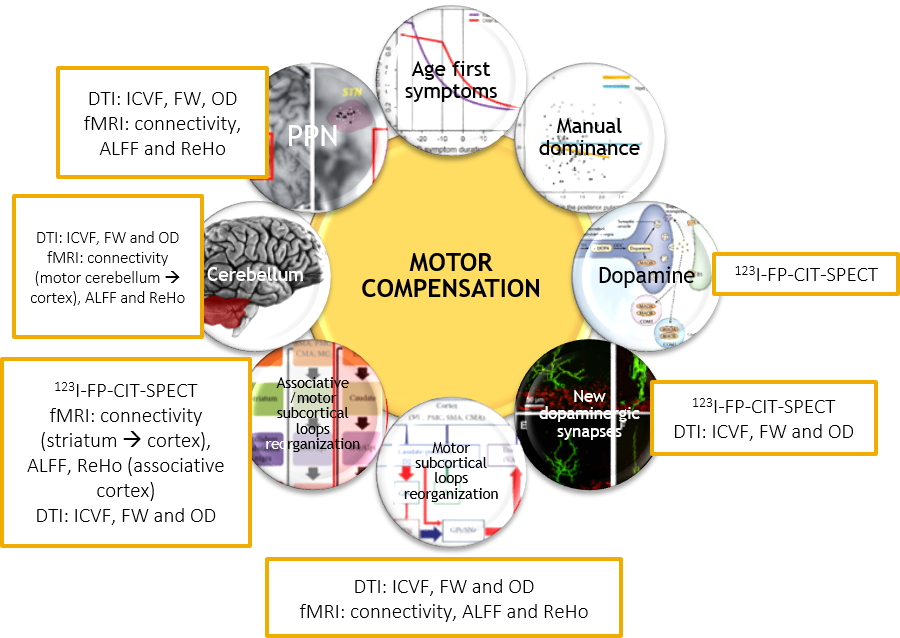
65 PD patients (<3 years after the diagnosis), 24 iRBD patients and 24 healthy controls were included in this study between 11/2014 and 10/2017. Every patient and subject underwent a large clinical assessment, a 123I-FP-CIT-SPECT (hybrid CT-SPECT Discovery 670 Pro GE™) and multi-modal 3T MRI (Siemens™ Magnetom FIT 3 tesla PRISMA) including 3DT1 MP2RAGE, DTI (3 acquisitions: 64 directions, 32 and 8 directions), T1 neuromelanin (voxel size: 0.4297x0.4297x3.0000mm3) and resting-state functional MRI (fMRI) acquisitions (under medication: 8min, 220 volumes). Briefly: SPECT data were corrected for Partial Volume Effects and segmented in striatal anatomical and functional subregions using the YeB atlas5. DTI data were transformed into OD, ICVF and FW maps using the NODDI algorithm. fMRI data were transformed into ReHo and ALFF maps and were also used for simple functional connectivity (CONN toolbox) and effective connectivity (spectral DCM) analyses. DTI and fMRI data were used either in the native (ROI) or MNI space (voxelwise). Neuromelanin alteration was quantified after manual SNpc segmentation using a hybrid intensity x volume measure. In order to assess the motor compensation mechanisms, we first created an adjusted motor severity index (aMSI), i.e. a ratio between the degree of akinetic motor severity (MDS-UPDRS akinetic subscore) and the degree of neuromelanin SNpc alteration: $$aMSI=((100+ZScoreMotor)/(100-ZScoreNeuromelanin)×100)-100$$ . Z-Scores were created according to a non-parametric approach: $$Z_i= (0.6745(x_i- (x ̃)))/MAD$$ where $$MAD= median{ |x_i - x ̃| }$$ and $$x ̃ = median of healthy controls$$. aMSI was then correlated to neuroimaging measurements defined according to our compensation hypotheses (Figure 1). Significant corrections were finally entered into a stepwise ascending multiple regression. Negative correlations were expected to reflect motor compensation mechanisms and positive correlations inefficient mechanisms. These correlations were performed amongst 68 patients (63 PD patients and 5 iRBD with significant 123I-FP-CIT-SPECT denervation within the sensorimotor putamen).RESULTS
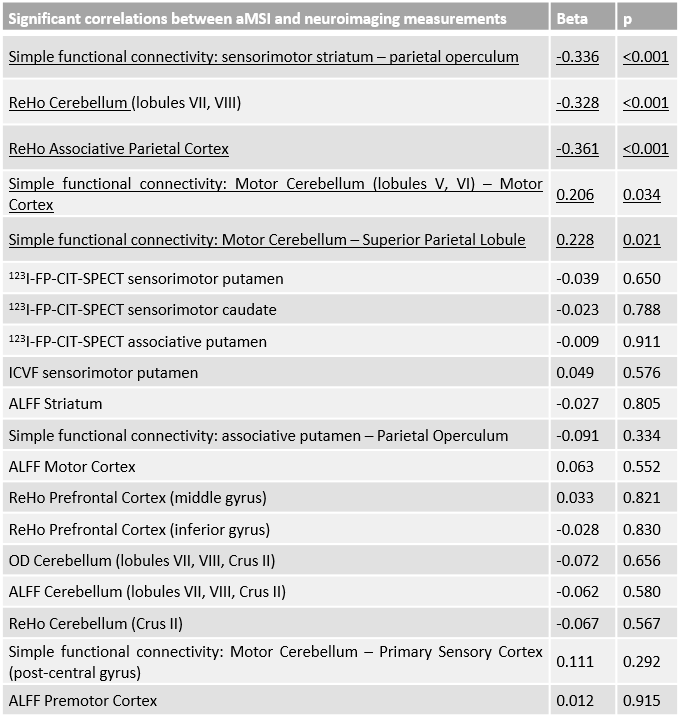
PD and iRBD patients had a significant decrease of 123I-FP-CIT-SPECT fixation within all the subareas of the striatum (with PD patients having a significant more severe loss of signal in every striatal subarea than iRBD patients). PD and iRBD patients had a significant alteration of the neuromelanin intensity x volume value without significant difference between the two groups (same results for intensity and volume independently). Significant imaging correlations with aMSI are listed in Figure 2. Amongst these significant correlations, the stepwise ascending multiple regression revealed that aMSI was negatively correlated to the simple functional connectivity between the sensorimotor striatum and the associative parietal cortex, the ReHo within the cerebellar lobules VII and VIII and within the associative parietal cortex. It was positively correlated to the simple functional connectivity between the cerebellar lobules V and VI and the primary motor cortex and to the simple functional connectivity between the cerebellar lobules V and VI and the superior parietal lobule (Figure 2).DISCUSSION
Amongst the tested compensation mechanisms in that study, the significant and independent factors that explain motor compensation in early PD patients is the reorganization of motor and cognitive subcortical loops and the reorganization of the associative areas of the cerebellum (lobules VII and VIII). On the other hand, the overuse of the motor cortical-cerebellum areas appears to be a marker of motor compensation failure. These compensation mechanisms might be underlain by preserved dopaminergic synapses within the striatum despite SNpc alteration (significant negative correlation between aMSI and 123I-FP-CIT-SPECT fixation within the sensorimotor striatum). Thus, new dopaminergic synapses or more active dopaminergic synapses are also probably a compensation mechanism in PD.CONCLUSION
New dopaminergic synapses or more active dopaminergic synapses, reorganization of motor and cognitive subcortical loops and of the associative areas of the cerebellum are the main motor compensation mechanisms in human early PD.Acknowledgements
No acknowledgement found.References
[1] Hornykiewicz, Wien. Klin. Wochenschr. 1963; 75: 309–12
[2] de La Fuente-Fernández et al., Ann. Neurol. 2011; 69: 803–810
[3] Bézard et al., Handb. Behav. Neurosci. 2010; 20: 641–652
[4] Helmich et al., Cereb. Cortex 2010; 20: 1175–1186
[5] Yelnik et al., NeuroImage 2007; 34: 618-638
Figures