2955
Functional cardiac MRI for monitoring progression of hypertrophic cardiomyopathy in Mybpc3 mouse modelsMin-Chi Ku1,2, Till Huelnhagen1, Saskia Schlossarek3,4, Andreas Pohlmann1, Lucie Carrier3,4, and Thoralf Niendorf1,2,5
1Berlin Ultrahigh Field Facility (B.U.F.F.), Max Delbrück Center for Molecular Medicine in the Helmholtz Association (MDC), Berlin, Germany, 2DZHK (German Centre for Cardiovascular Research), partner site Berlin, Berlin, Germany, 3Department of Experimental Pharmacology and Toxicology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany, 4DZHK (German Centre for Cardiovascular Research), partner site Hamburg/Kiel/Lübeck, Hamburg, Germany, 5Experimental and Clinical Research Center, Charite Medical Faculty and the Max Delbrueck Center for Molecular Medicine in the Helmholtz Association, Berlin, Germany
Synopsis
Mutations in gene MYBPC3, encoding cardiac myosin-binding protein C, cause hypertrophic cardiomyopathy (HCM), which is characterized by left ventricular hypertrophy (LVH), diastolic dysfunction, increased interstitial fibrosis, and may lead to sudden cardiac death and heart failure. In spite of the advances in translational medicine, we know very little about HCM. The HCM progression is complex and shows heterogeneous phenotypes. The missing linkage of in vivo imaging and pathology has hindered the investigation of detail mechanisms of HCM. We therefore investigated Mybpc3-targeted mouse models using CMR markers for understanding HCM pathophysiology and to get closer to complete pictures of HCM progression.
Introduction
HCM is one of the most frequent cause of death in young athletes, and the main cause of heart failure at any age. The prevalence of HCM is 1:200 [1]. Its etiology is unknown, but ~60% of HCM patients carry genetic variants in sarcomere proteins. Among them, MYBPC3 accounts for 50% of genotyped patients. MYBPC3 is expressed exclusively in the myocardium, where it associates with the sarcomere filament, provides structural support and regulates both contraction and relaxation [2]. Despite the substantial advances in translational medicine, diagnosis of HCM relies only on the identification of left ventricular hypertrophy (LVH), which is a symptom of advanced disease progression, through echocardiography or cardiac MRI (CMR). However, HCM progression is complex and involves intracellular and extracellular alterations that manifest clinically as changes in mass, geometry and function of myocardium. The absence of connection between in vivo imaging and pathophysiology hindered the investigation of the detailed mechanisms of HCM. For monitoring HCM progression non-invasive imaging alone is not sufficient for phenotyping. Here we aim for investigating Mybpc3-targeted mouse models using CMR markers for understanding HCM pathophysiological mechanisms.Methods
We used two models which bear key features of HCM. One is a murine model (DBA2J/D2 strain) that naturally carries genetic variants in Mybpc3 [3]. The second model is a Mybpc3-targeted knock-in (KI) mouse [4]. Three month old male D2 (n=4) and reference strain C57BL/6 (B6; n=3), heterozygous (HET, n=3) and homozygous (KI, n=3) mice were used for in vivo CMR. A 9.4T animal MR scanner (Biospec 94/20) and a cryogenic transceiver RF coil (CryoProbe, all from Bruker Biospin, Germany) were used. To obtain cardiac short axis (SAX) views covering the whole heart, ten slices were consecutively acquired using self-gated FLASH (IntraGate, TE/TR= 1.58/8.5ms, FA = 20°, BW = 98kHz, FOV=11×22mm2, matrix=192×384, thickness=0.8mm, cardiac frames=16) [5]. Functional assessments were performed using CMR42 (Circle CVI, Canada) and analyzed on a slice-by-slice basis. Endo- and epi-cardiac borders were manually segmented. Ejection fractions (EF) and myocardial masses were calculated for LV and RV. After in vivo CMR, mice were perfused with 4% PFA. For detecting early fibrosis and distinguish different forms, Sirius Red (stain for collagen) was stained on paraffin sections.Results
For the natural HCM model, LVH was significantly higher in D2 than in B6 mice (LVwall thickness=1.27±0.06mm vs 0.99±0.04mm, P<0.01). Whereas LVEF did not decrease in D2 mice (D2: 70.2±5.7% vs B6: 73.8±0.7%), RVEF was lower in D2 than B6 mice (61.6±7.9% vs 74.9±1.5%, P<0.05; Fig. 1). In human, most of the known MYBPC3 defects are truncating mutations, which are recapitulated in Mybpc3-targeted KI mice. After careful analysis of the CINE images (Fig. 2A), we found that KI mice developed severe LVH compared to WT mice (LVwall thickness=1.34±0.13mm vs 1.01±0.04mm, P<0.01) and severe global functional impairment in both LV (LVEF=39.2±5.9% vs 80.6±8.9%, P<0.01) and RV (RVEF=37.4±9.0% vs 70.3±3.7%, P<0.01). In contrast to homozygous KI mice, HET mice did not develop LVH but exhibited trends towards increased heart mass. LVEF did not differ between HET and WT mice (74.2±1.3% vs 80.6±8.9%), whereas RVEF showed a tendency towards lower values in HET (62.5±4.9% vs 70.3±3.7%; Fig. 2B). Sirius Red staining revealed a normal amount of perivascular collagen deposition in WT mice, slightly diffused fibrosis in HET mice and severe fibrosis in KI mice (Fig. 3).Discussion and Conclusions
We characterized the phenotypic impact of Mybpc3 mutations on cardiac structure and function with CMR. HCM mice developed profound LVH, the main phenotype of HCM, and RV dysfunction. The advantage of using genetic mouse models is that we can follow the disease progression, and decipher the genetic influence of HCM. For example, the absence of LVH and cardiac dysfunction in young HET mice mimics the situation in young individuals carrying this heterozygous HCM mutation. Interestingly, the slight decrease in RV function depicted by CMR and the mild interstitial fibrosis showed by histological assessment in HET mice indicate that subtle cardiac functional change might be independent or may occur prior to LVH. Furthermore, given that RV involvements may be comparable to LV function impairments in HCM patients, the presence of RV dysfunction captured by CMR may predict the severe symptomatic HCM and help to study the HCM progression and symptom occurrence. In conclusion, these data suggest hints for the development of a predictable imaging biomarker ultimately for early probing of HCM. In addition, efforts on in vivo CMR should focus on detecting adverse phenotypes, so that interventional treatment strategies can be initiated earlier in the clinical course for HCM treatment.Acknowledgements
No acknowledgement found.References
[1] Semsarian C et al. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015; 65:1249. [2] Carrier L et al. Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene, 2015; 573:188 [3] Zhao W et al., A Murine Hypertrophic Cardiomyopathy Model: The DBA/2J Strain. PLoS One. 2015;10:e0133132. [4] Vignier N et al. Nonsense-Mediated mRNA Decay and Ubiquitin–Proteasome System Regulate Cardiac Myosin-Binding Protein C Mutant Levels in Cardiomyopathic Mice. Circ Res. 2009;105:239. [5] Wagenhaus B et al., Functional and Morphological Cardiac Magnetic Resonance Imaging of Mice Using a Cryogenic Quadrature Radiofrequency Coil. PLoS One. 2012;7:e42383.Figures
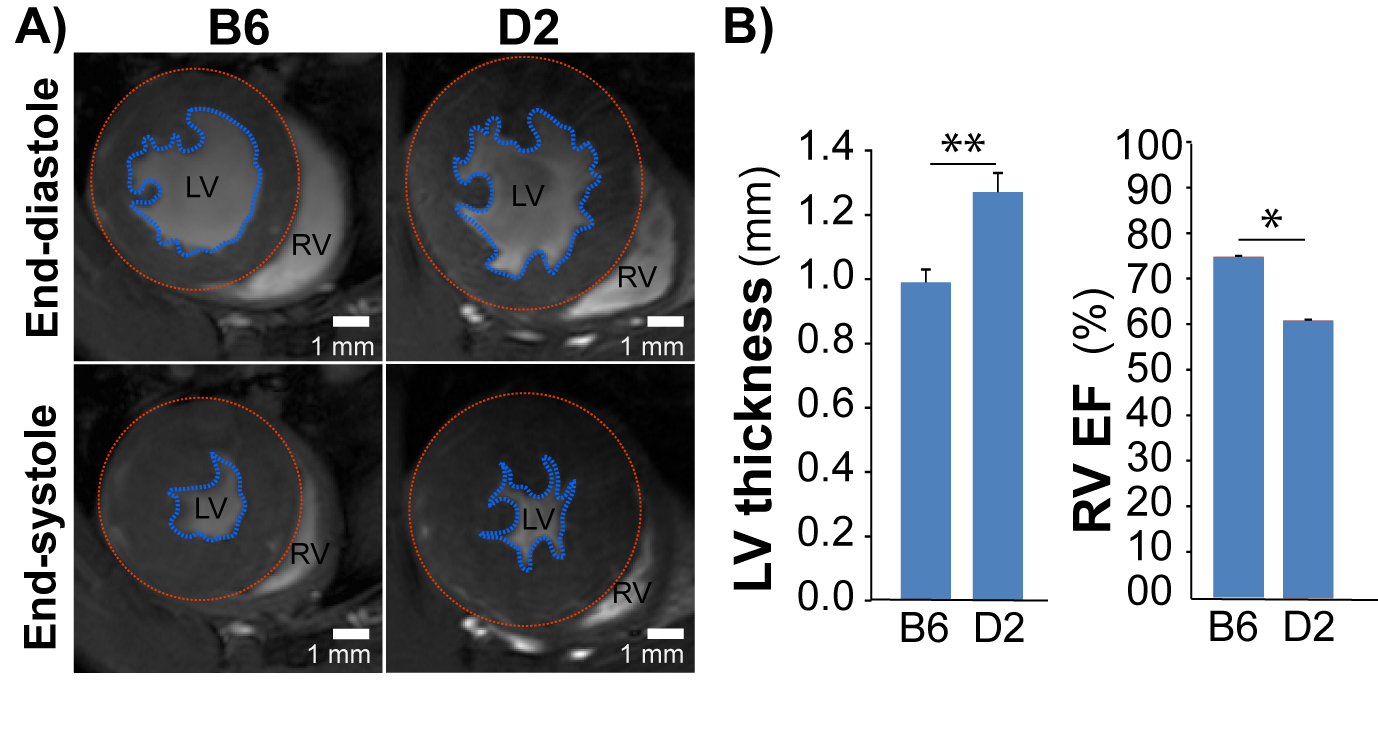
Fig 1: HCM mice
naturally carry Mybpc3 gene variant show structural and
functional changes. A), Representative FLASH CINE images (upper
row: end-diastole and lower row: end-systole) show mid-ventricular SAX views in
male B6 (C57BL/6) and D2 (DBA/2J) mice hearts. Blue contours outline the endocardium,
red contours outline the epicardium. B), Functional
assessments by CMR shows reduced RV function in D2 HCM mice (n=4) compared to
B6 (n=3). *p<0.05; **p<0.01
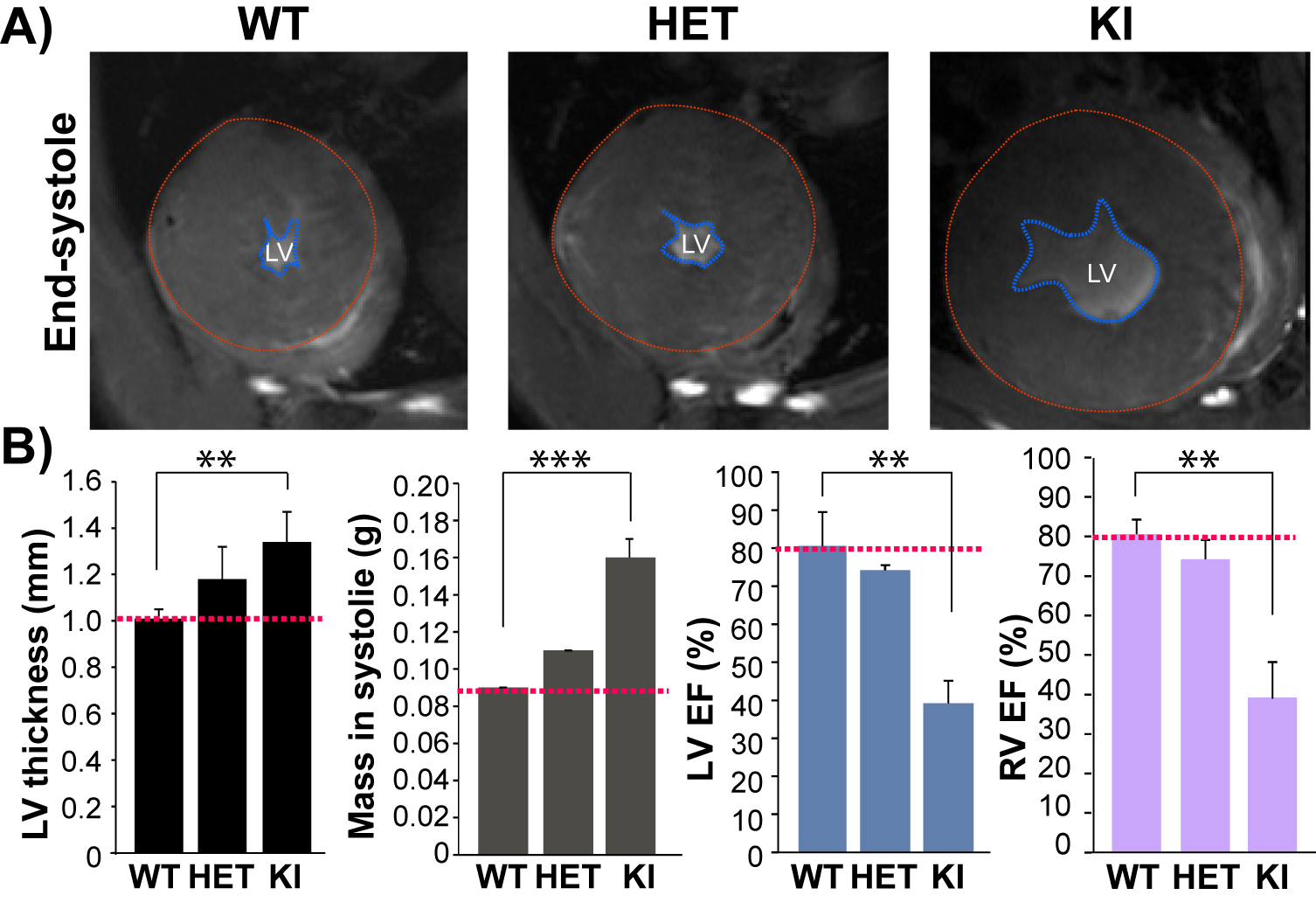
Fig. 2: HCM mice genetically
targeted in Mybpc3 gene show structural
and functional changes. A), Representative FLASH CINE images
(end-systole) show mid-ventricular SAX views in male WT, HET, and KI mice
heart. Blue dash lines outline the endocardium. Red
dash lines outline the epicardium. B), Functional assessments by CMR show defected LV and RV function
in homozygous KI mice (n=3) compared to WT (n=3). **p<0.01; ***p<0.001
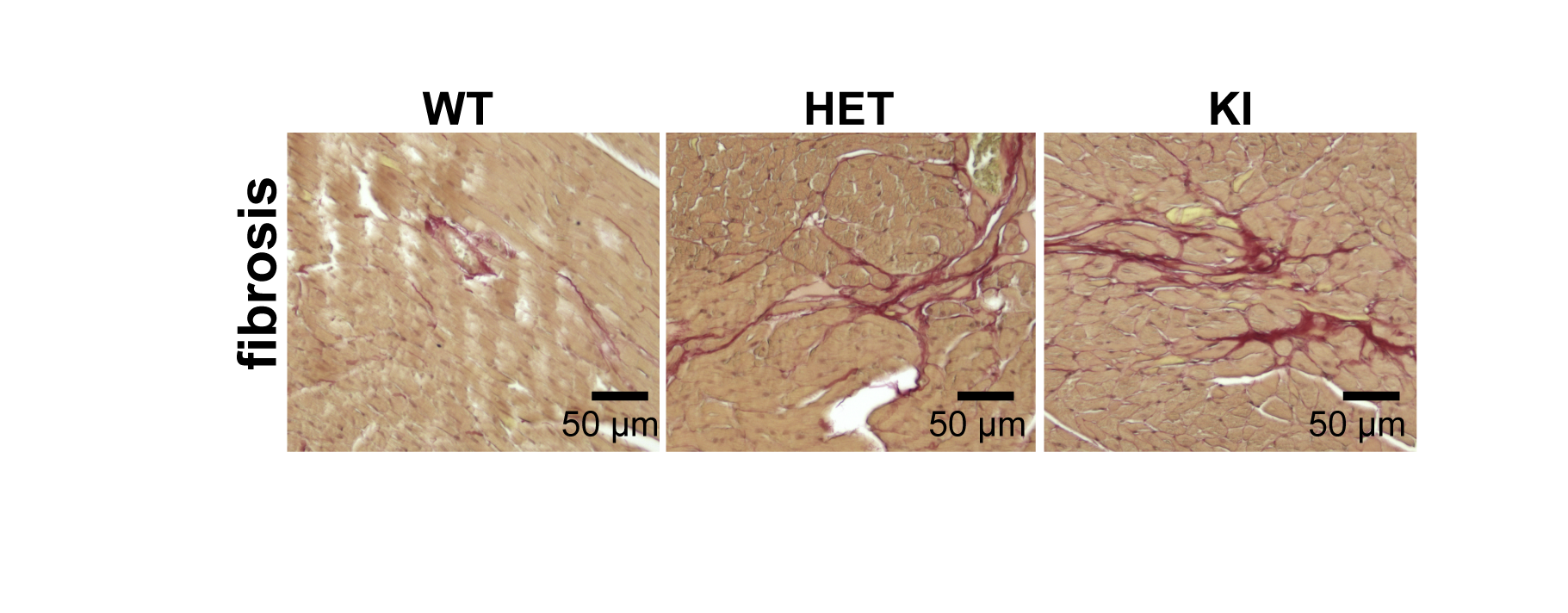
Fig. 3: Mice genetically targeted in Mybpc3 gene show myocardial fibrosis. Representative color images of mice heart sections stained with Sirius Red in male WT, HET, and KI heart tissue. WT heart presents the normal amount of perivascular collagen deposition. HET heart shows mild fibrosis and KI heart shows more severe fibrosis.