2032
Effects of perivascular progenitor cells in combination with Abeta clearance on neurovascular function following transient hypertension in a transgenic rat model of Alzheimer’s Disease1Sunnybrook Research Institute, Toronto, ON, Canada, 2Medical Biophysics, University of Toronto, Toronto, ON, Canada, 3CReATe Research Program, Toronto, ON, Canada, 4Laboratory Medicine and Pathobiology, University of Toronto, Toronto, ON, Canada
Synopsis
Examining the interplay between cerebrovascular compromise and AD in the development of therapies is complicated by long prodromal phases of both conditions, necessitating preclinical studies. Four-month-old TgAD-F344 rats, which by six months of age present amyloid deposits and hyperphosphorylated tau, were treated with a nitric oxide synthase inhibitor L-NAME for one month to induce transient hypertension. Human umbilical cord perivascular cells were then given in combination with scyllo-inositol, an inhibitor of Abeta peptide oligomerization and fibrillization to elicit cerebrovascular repair and clear amyloid. Following L-NAME, non-transgenic rats showed transient cerebrovascular changes, whereas TgAD-F344 animals exhibited sustained increase in cerebrovascular reactivity. The latter effect was ameliorated by the treatment.
Introduction
Examining the interplay between cerebrovascular compromise and Alzheimer’s disease and development of therapeutic interventions is complicated by long prodromal phases of both conditions, necessitating preclinical studies. Our therapeutic approach is designed to target sporadic AD patients with established AD pathophysiology in combination with vascular injury. We combined a model of transient hypertension (via administration of NOS inhibitor Nω-nitro-L-arginine methyl ester hydrochloride, L-NAME) with a transgenic rat model that recapitulates Aβ peptide deposition and tau hyperphosphorylation in the adulthood followed by cognitive decline and neuronal dysfunction and loss [1-3]. We administered human umbilical cord perivascular cells (HUCPVC) to elicit brain vascular repair and scyllo-inositol (SI) to clear amyloid. Treatment outcomes were measured by continuous arterial spin labelling (CASL), immunohistochemistry (inflammatory markers, amyloid), and immunoblotting (nitric oxide synthases, markers of vascular remodeling).Methods
Four-month-old TgAD-F344 rats were treated with the partially selective NOS inhibitor L-NAME to induce transient hypertension. Following return to normotension after cessation of L-NAME, immunosuppression with cyclosporine A and SI treatment were initiated one week prior to HUCPVC transplantation. HUCPVC treatment was given four-weeks post L-NAME treatment, and final imaging session conducted four weeks thereafter. CASL was performed during both rest and CO2 challenges at: baseline, immediately post-L-NAME treatment, and at endpoint to measure cerebrovascular reactivity changes over time. Imaging was performed using a 7T horizontal preclinical MRI system (Bruker BioSpec). Animals were intubated, mechanically ventilated, and challenged by four presentations of a hypercapnic mixture in ON:OFF periods of 1:4 minutes (ON: 10% CO2 , 30% O2 and 60% N2 , OFF: 0% CO2 , 30% O2 and 70% N2 ). Inspired mixture composition and delivery were controlled by a programmable GasMixer (GSM3, CWE Inc., Boston MA). Using a 1.5-second adiabatic labelling pulse, and a 0.4-second post-labelling delay, single average, single shot echoplanar images (EPI) were obtained from two 1.5mm thick coronal slices positioned over the sensorimotor cortex, with a 0.25x0.25mm 2 in-plane resolution, TR/TE of 2000/8.3ms, and interslice gap of 0.5mm, following our previous work [4]. Changes in CASL signal were assessed via AFNI’s 3dDeconvolve.Results
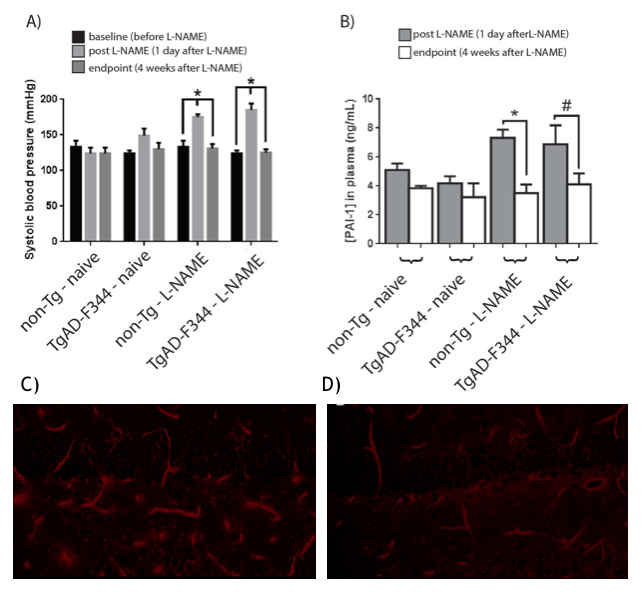
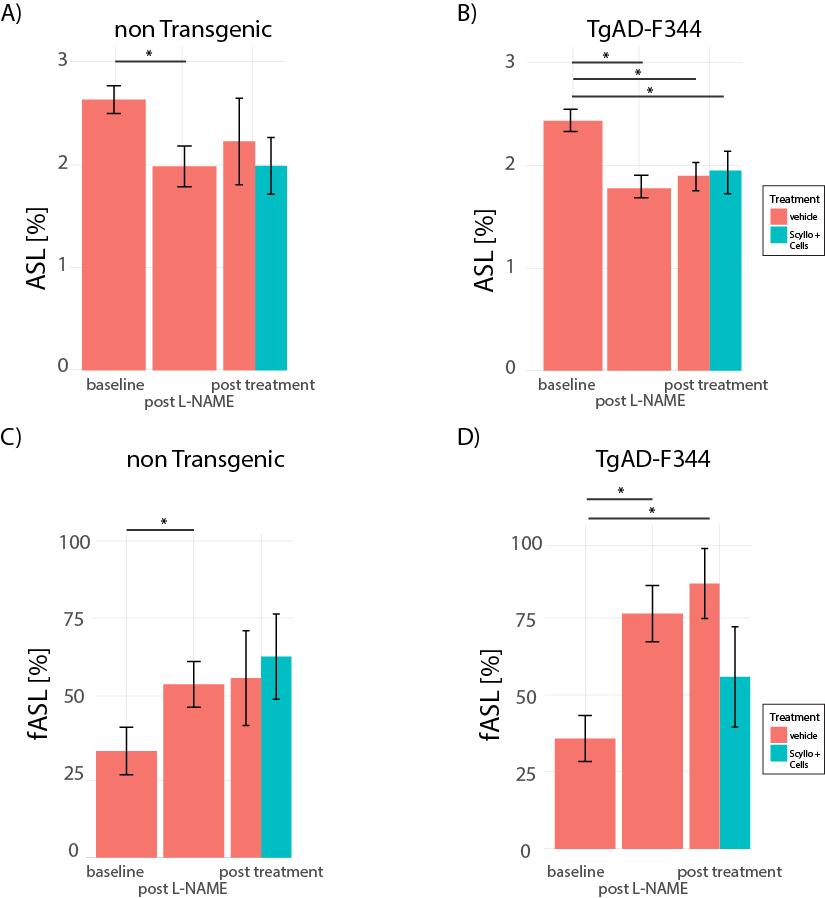
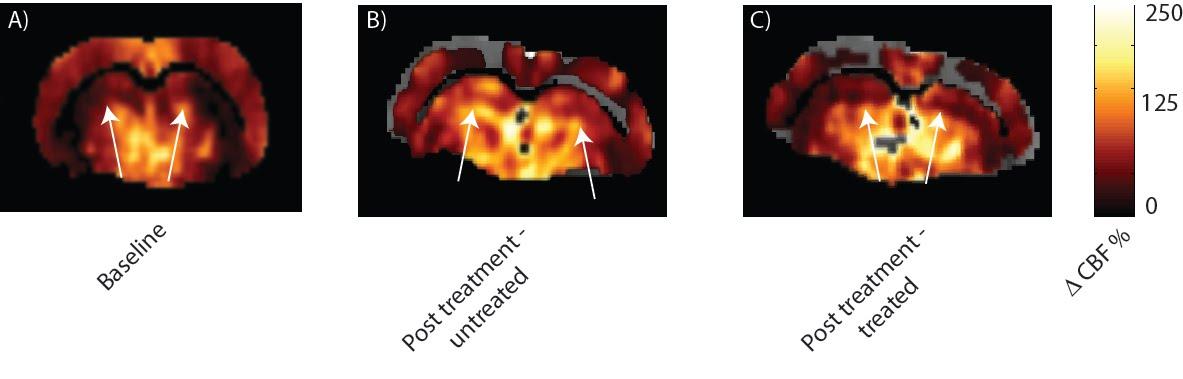
L-NAME administration induced moderate hypertension in both TgAD-F344 and non-TgAD rats, manifested by increased systolic blood pressure (Figure 1A) and elevated plasma level of plasminogen activator inhibitor-1 (PAI-1) (Figure 1B), which was accompanied by decreased vessel density on lectin staining of the endothelium (Fig1 C: baseline; Fig 1D: post-L-NAME) and a ~25% reduction in resting hippocampal perfusion (Figure 2A, 2B post-L-NAME vs. baseline ASL p<0.05). In the non-TgAD, hippocampal perfusion returned to baseline levels 4-weeks post cessation of L-NAME (Figure 2A endpoint vs. baseline ASL), whereas in TgAD-F344 rats hippocampal hypoperfusion persisted until the endpoint (Figure 2B, endpoint vs. baseline ASL p<0.05). Combined treatment of scyllo-inositol and HUCPVC did not affect resting hippocampal CBF in the TgAD-F344: they showed persistent hippocampal hypoperfusion at the end of the treatment (Figure 2B endpoint vs. baseline ASL, p<0.05). Hippocampal vascular responsivity to hypercapnia was increased (by ~60% in non-TgAD and by ~120% in TgAD-F344) following L-NAME administration (Figure 2C and 2D post-L-NAME vs. baseline fASL, p<0.05) in both non-TgAD and TgAD-F344 animals, but it remained elevated over the next 2 months only in the vehicle-administered TgAD-F344 rats (Figure 2D endpoint vehicle-administered vs. baseline fASL, p<0.05). Combinatorial treatment of TgAD-F344 animals rescued their hippocampal responses to hypercapnia to baseline levels (Figure 2D, endpoint treated vs. baseline fASL). Representative CASL images of cerebrovascular reactivity in TgAD-F344 rats at baseline and post-vehicle or post-SI+HUCPVC treatment are shown in Figure 3. In addition, treated TgAD-F344 animals showed reduced hippocampal amyloid load when compared to that in vehicle administered TgAD-F344 animals.Conclusions
Transient hypertension caused sustained cerebrovascular damage in a transgenic model of Alzheimer’s disease, with decreased resting hippocampal perfusion and elevated hippocampal vascular responsivity to hypercapnia, revealing an interaction between transient hypertension and AD pathology. Treatment with scyllo-inositol and HUCPVC normalized hippocampal vascular responses to hypercapnia but not the resting perfusion. Combinatorial treatment paradigms may be necessary to address AD with comorbid vascular disease.Acknowledgements
No acknowledgement found.References
1. Cohen, R.M., et al., A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric abeta, and frank neuronal loss. J Neurosci, 2013. 33(15): p. 6245-56. 2. Joo, I.L., et al., Early neurovascular dysfunction in a transgenic rat model of Alzheimer's disease. Sci Rep, 2017. 7: p. 46427. 3. Bazzigaluppi, P., et al., Early-stage attenuation of phase-amplitude coupling in the hippocampus and medial prefrontal cortex in a transgenic rat model of Alzheimer's disease. J Neurochem, 2017. 4. Lake, E.M., et al., Neurovascular unit remodelling in the subacute stage of stroke recovery. Neuroimage, 2017. 146: p. 869-882.Figures