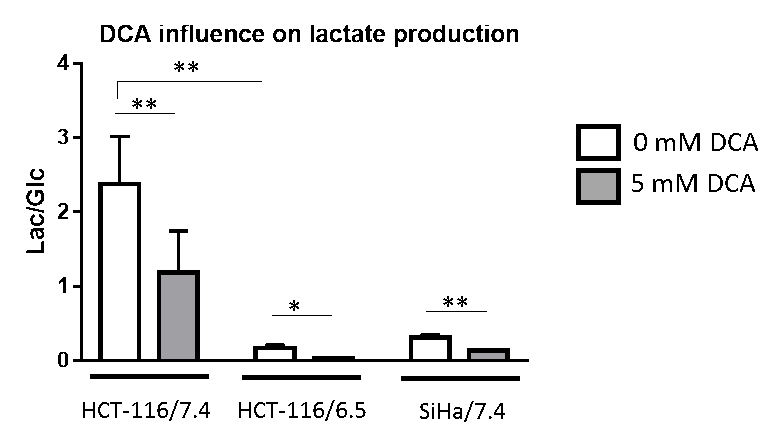1341
13C-NMR to study cancer cell metabolic plasticity following PDK inhibition. Influence of dichloroacetate and long-term exposure to acidic environment on glucose and glutamine metabolic pathways.1Biomedical Magnetic Resonance Group (REMA), Louvain Drug Research Institute, Catholic university of Louvain, Bruxelles, Belgium, 2Pharmacotherapy Group (FATH), Institute of Experimental and Clinical Research, Catholic university of Louvain, Bruxelles, Belgium
Synopsis
Many cancer cells present an exacerbated glycolytic flux that provides advantage for growth and leads to extracellular acidosis. Dichloroacetate (DCA), a PDK inhibitor, shifts metabolism from glycolysis to glucose oxidation and decrease various cancer cells lines proliferation. However, as tumor cells are presenting metabolic plasticity, PDK inhibition may lack efficacy. To measure metabolic adaptations of cancer cells to acidic environment and in response to DCA, we studied metabolic fluxes using 13C-NMR spectroscopy. With this technology, we measured differences in metabolic profiles between parental cancer cells line and acidic clones and we quantified specific changes in metabolism following DCA treatment.
Introduction
Reprogramming energy metabolism is an emerging target of cancer. Glycolysis is frequently upregulated in highly proliferative cancer cells because it provides biosynthetic advantage for growth. Dichloroacetate (DCA), a PDK inhibitor currently tested in the clinics, is known to shift metabolism from glycolysis to glucose oxidation and to decrease the proliferation in various cancer cells lines1. However, as tumor cells are presenting extraordinary metabolic plasticity, PDK inhibition may lack efficacy because of the induction of compensatory mechanisms sustaining tumor growth. Moreover, this exacerbated glycolytic flux in cancer cells leads to extracellular tumor acidosis2. Conversely, little is known about how tumor cells adapt their metabolism to acidosis. Our hypothesis is that glutamine may induce alternative metabolic routes when using PDK inhibitors. Glutamine is an important amino acid that can support cell proliferation3. Furthermore, long-term exposure of cancer cells to acidic pH leads to a metabolic reprogramming toward glutamine metabolism3. To measure metabolic adaptations of cancer cells to acidic environment and in response to DCA, we studied metabolic fluxes using 13C-NMR spectroscopy. This will provide a strong rationale for the association of treatments to be used in combination with PDK inhibitors.Methods
Cancer cell lines with different metabolic profiles (SiHa; oxidative, HCT-116; glycolytic) and their pH-acidic adapted clones were incubated with +/- 5 mM of DCA and one 13C tracer (glucose-13C6 or glutamine-5-13C). After 24h, extracellular media were collected and polar cellular metabolites were extracted. 13C-NMR spectra of cellular extracts and media were acquired on a 600 MHz Bruker NMR equipped with a cryoprobe. The acquisition time was 0.8 s with 1024 repetitions and 10 s of interpulse delay. Spectrum analysis, assignment and quantification were performed with the Amix software. We also assessed, by proliferation assay, the toxicity of DCA in each cell lines and their acidic clones.Results
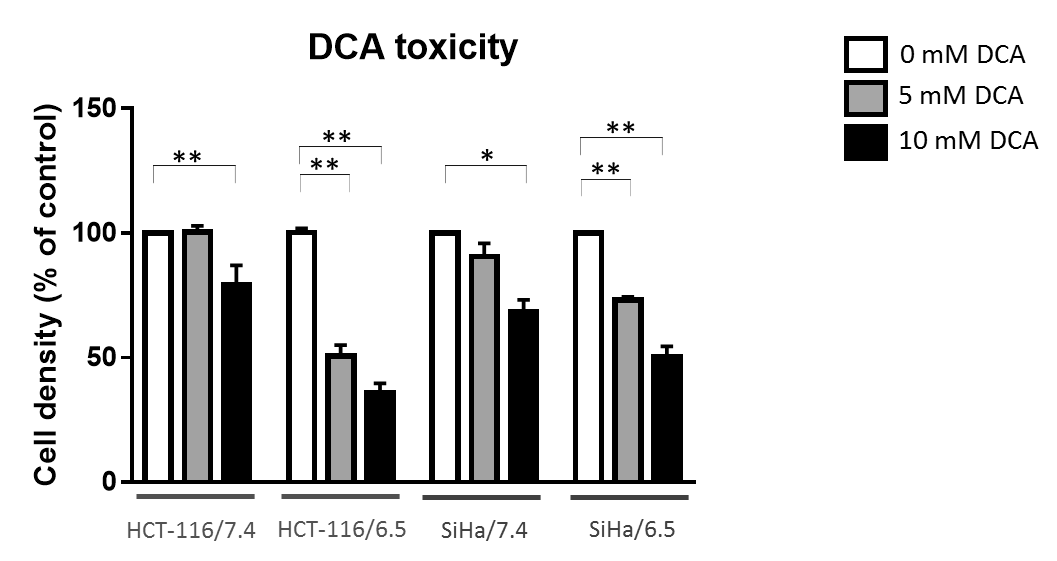
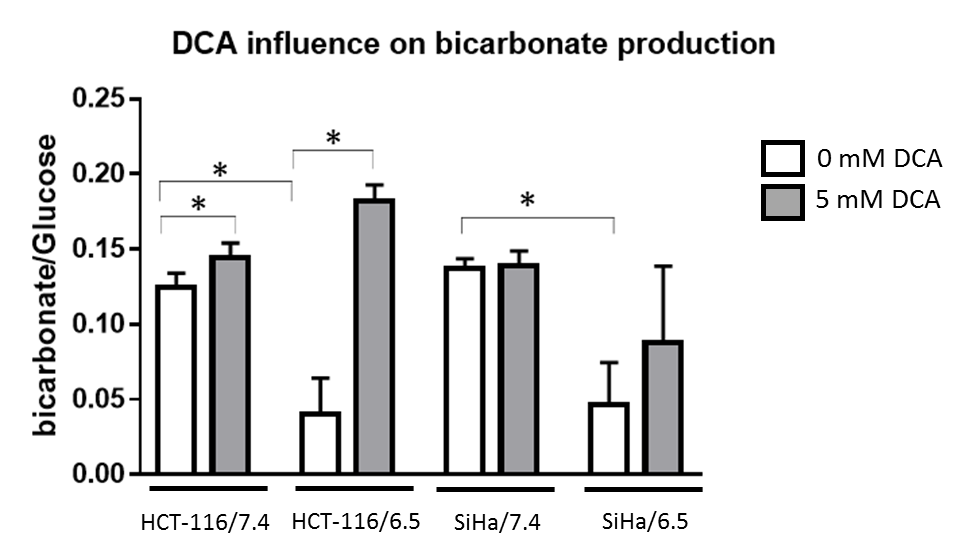
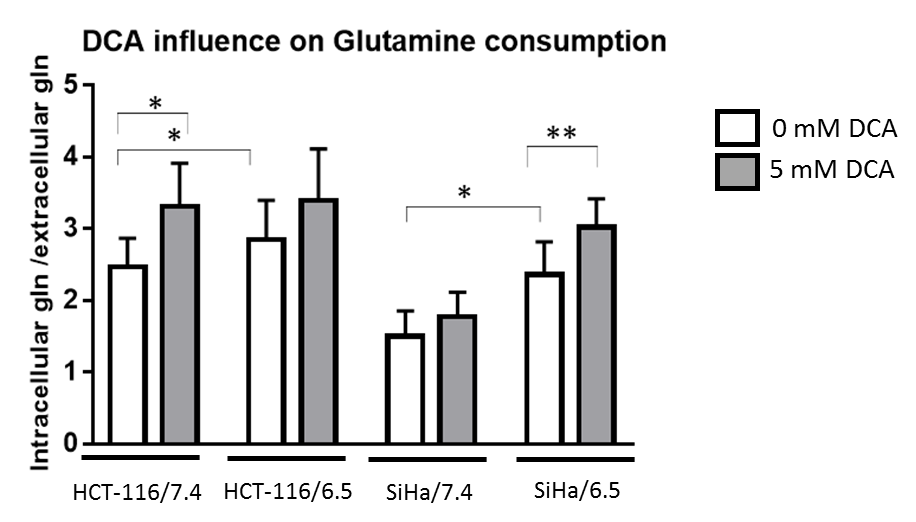
DCA induced more toxicity in HCT-116 and SiHa acidic clones compared to the parental cell lines (fig.1). The production of intracellular lactate from glucose-13C6 in HCT-116 was 90± 2% lower in the acidic clones than the parental cells (fig.2). We detected no lactate in the SiHa acidic clones. DCA decreased the intracellular lactate production from glucose of 57± 7% in HCT-116 parental cells, of 83± 3% in HCT-116 acidic clones and of 54± 4% in SiHa parental cells (fig.2). Similar results were found for extracellular lactate. DCA inhibited the conversion of pyruvate to acetyl-CoA and CO2, this last being converted into bicarbonate, quantifiable in extracellular media spectra. Figure 3 shows that the production of bicarbonate from glucose-13C6 was lower in the acidic clones than in the parental cells (lower of 68± 23% for HCT-116 and 67± 20% for SiHa). DCA increased bicarbonate production of 17± 3% in HCT-116 parental cells and of 356%± 28% in their acidic clones (fig3). DCA had no significant effect on SiHa parental and acidic clones. DCA also decreased alanine production and pyruvate export in HCT-116 parental cell lines, these metabolites were not detectable in the other cell lines. Figure 4 shows that basal glutamine-5-13C consumption was higher in acidic clones than in parental cell lines (14± 3% for HCT-116 and 69± 29% for SiHa). In the presence of DCA, glutamine consumption was increased in HCT-116 parental and acidic cells and in SiHa parental and acidic cells (33± 8%, 18± 7%, 25±17% and 32± 9% respectively). Interestingly, DCA decreased glutamate production from glutamine in all cell lines and increased aspartate production from glutamine in both parental cell lines. Aspartate was not detectable in acidic clones.Discussion
Although the toxicity of DCA was more important in acidic clones, they adapted their metabolism to be less dependent on glucose than parental cells (lower lactate and bicarbonate production) and more dependent of glutamine metabolism (more glutamine consumption). Metabolic modulation of DCA was dependent of the cell type and acidic environment. Glutamine consumption was increased in the presence of DCA suggesting a compensatory mechanism. Interestingly, in the presence of DCA, glutamate production was decreased but aspartate production was increased. These observations warrant further investigation to understand to role of glutamine pathway in the response of tumor cells to DCA treatment.Conclusion
Using 13C-NMR spectroscopy allow us to study cancer cell metabolic adaptation following PDK inhibition and under acidosis.Acknowledgements
No acknowledgement found.References
1. Bonnet, Sébastien, et al. "A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth." Cancer cell 11.1 (2007): 37-51.
2. Corbet, Cyril, et al. "The SIRT1/HIF2α axis drives reductive glutamine metabolism under chronic acidosis and alters tumor response to therapy." Cancer research 74.19 (2014): 5507-5519.
3. Corbet, Cyril, and Olivier Feron. "Metabolic and mind shifts: from glucose to glutamine and acetate addictions in cancer." Current Opinion in Clinical Nutrition & Metabolic Care 18.4 (2015): 346-353.
Figures