0450
Human Glioblastoma Cell Lines Co-oxidize [2,4-13C]betahydroxy-butyrate and [U-13C]-glucose: A 13C NMR Spectroscopic StudyOmkar B. Ijare1, Athena Hoppe1, Cole Holan1, Martyn A Sharpe1, David S Baskin1, and Kumar Pichumani1
1Kenneth R. Peak Center, Department of Neurosurgery, Houston Methodist Research Institute, Houston, TX, United States
Synopsis
Ketogenic diet has been
proposed for the adjuvant therapy in the treatment of brain tumors. The
rationale behind using ketogenic diet in the cancer treatment is the inability
of tumor mitochondria to oxidize KBs. Recent studies on ketone body metabolism
using 9L and RG2 cell lines and glioma models suggest that brain tumor
mitochondria are capable of oxidizing ketone bodies. The current study was undertaken
to determine relative utilization of betahydroxy-butyrate (BHB) and glucose in patient-derived
glioblastoma cells. Our findings clearly indicate that human brain tumor cells are fully capable of oxidizing ketone bodies even under
normoglycemic conditions.
Introduction
Warburg hypothesized that tumor cells have defective mitochondria and they heavily depend on glycolysis for their energy needs. It is well-known that normal brain cells metabolize ketone bodies (KBs) under hypoglycemic conditions. It has been postulated that brain tumor cells lack the ability to metabolize KBs. Based on above hypotheses, ketogenic diet (KD) has been proposed for the adjuvant therapy in the treatment of brain tumors [1]. KD, composed of high fat, low carbohydrate, and moderate protein, produces high levels of KBs in the circulation, and it is thought that defective mitochondria would not metabolize KBs and tumor cells would starve to death. Recent studies performed in rat cell lines/glioma models revealed that some tumor cells are capable of oxidizing KBs [2, 3]. These results contradict the hypothesis that brain tumors lack the ability to metabolize KBs. Cerebral ketone body metabolism differs between rodents and humans [4]. In the current study, we are investigating simultaneous oxidation of ketone body (betahydroxy-butyrate, BHB) and glucose in patient-derived glioblastoma multiforme (GBM) cell lines using 13C-NMR based isotopomer analysis.Methods
GBM tumor tissues were collected from patients undergoing craniotomy at the Houston Methodist Hospital following an approved IRB protocol. Cells were extracted from the tumor tissue and were initially grown in Dulbecco’s modified Eagle’s medium (DMEM) incubated at 37°C under humidified air with 5% CO2. Final 3 hours, the cells were grown in the DMEM media containing 4 mM [2,4-13C]betahydroxy-butyrate ([2,4-13C]BHB) and 6.0 mM [U-13C]glucose. After 3 hours, the media was removed, cells were washed with PBS buffer and the cell pellets were snap-frozen in liquid nitrogen. Prior to NMR data collection, the cell pellets were extracted in 5% perchloric acid, extracts were lyophilized and was reconstituted in 180 µL D2O containing 1.0 mM DSS-d6 (pH = 7.4). The sample-solution was transferred to a 3-mm NMR tube, and 1H and 1H-decoupled 13C NMR spectra were collected on a Bruker 600 MHz spectrometer equipped with a 5-mm cryo-probe optimized for direct 13C detection.Results
Figure 1 is a schematic showing metabolism of [2,4-13C]BHB and [U-13C]glucose in a tumor cell. [2,4-13C]BHB is converted to [2,4-13C]acetoacetate by the enzyme BHB dehydrogenase, and further metabolized to [2-13C]acetyl-CoA. On the other hand, [U-13C]glucose is converted to [U-13C]pyruvate via glycolysis which generates [1,2-13C]acetyl-CoA through pyruvate dehydrogenase. [2-13C]acetyl-CoA enters the TCA cycle, condenses with unlabeled oxaloacetate to form [4-13C] labeled α-ketoglutarate (α-KG) and glutamate in the first turn of the cycle. After multiple turns of the cycle, carbons 3 and 4 of α-KG and glutamate are 13C-labeled. [U-13C]glucose-derived [1,2-13C]acetyl-CoA, in the first turn of the TCA cycle, forms 4- and 5- 13C-labeled α-KG and glutamate. After multiple turns of the cycle, 3, 4, and 5 carbons of α-KG and glutamate are 13C-labeled. Figure 2 shows the portion of the 13C NMR spectrum of C4 signal of glutamate with various multiplicities. The singlet (S) and the doublet (D34) are due to [4-13C]glutamate and [3,4-13C]glutamate respectively, which are derived from the metabolism of [2,4-13C]BHB. Doublet (D45) and the quartet (Q) are due [4,5-13C]glutamate, and [3,4,5-13C]glutamate respectively, which are derived from [U-13C]glucose. Figure 3 shows a chart of normalized peak areas of S, D34, D45 and Q peaks of C4-glutamate signal. The relative contribution of BHB and glucose to C4 glutamate signal is determined as follows: In C4 glutamate carbon multiplets, the sum of S and D34 is a measure of [2,4-13C]BHB, whereas the sum of D45 and Q is a measure of [U-13C]glucose. From the 13C-isotomer analysis, we found that the relative contributions of [2,4-13C]BHB and [U-13C]glucose to the C4 glutamate 13C fractional enrichment were 42.60% and 57.40% respectively.Discussion
KDs are under clinical trials [1] for the adjuvant therapy for the treatment of brain tumors. The rationale behind using KDs in the cancer treatment is the inability of tumor mitochondria to oxidize KBs. Ketone body metabolism in rat cell lines/glioma models [2,3] revealed that brain tumor mitochondria are capable of oxidizing KB. Results from the current study using patient-derived tumor cell lines clearly indicate that human GBMs are fully capable of oxidizing KB in the presence of glucose.Conclusion
Our findings show that human GBM oxidize BHB even under normoglycemic conditions.Acknowledgements
This study was supported by the Donna and Kenneth R. Peak Foundation, The Kenneth R. Peak Brain and Pituitary Tumor Treatment Center at Houston Methodist Hospital, The Taub Foundation, The John S. Dunn Foundation, The Blanche Green Estate Fund of the Pauline Sterne Wolff Memorial Foundation, The Verelan Foundation, The Houston Methodist Hospital Foundation, The American Brain Tumor Association, and by many brave patients and families who have been impacted by the devastating effects of brain cancers and central nervous system disease. We thank Sophie Lopez for helping with cell culture experiments.References
- https://clinicaltrials.gov/ct2/show/NCT03160599 (Accessed November 8, 2017)
- Feyter HM, Behar KL, Rao JU et al., Neuro-Oncology, (2016) 18:1079- 1087.
- Eloqayli H, Melo TM, Haukvik A, Sonnewald U. Neurochem Res (2011) 36:1566-1573.
- Morris AA. J Inherit Metab Dis. (2005) 28:109-121.
Figures
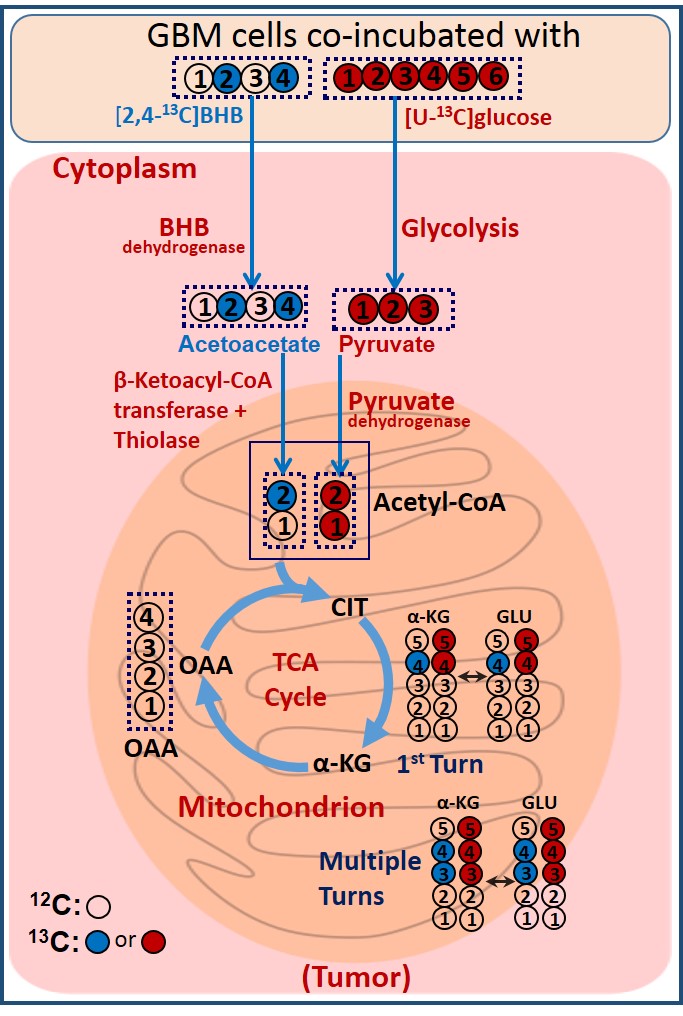
Schematic illustrating simultaneous oxidation of [2,4-13C]BHB
and [U-13C]glucose in a brain tumor cell. 13C-labeling in
α-KG and glutamate arising from [2,4-13C]BHB and [U-13C]glucose have been depicted
by blue and red filled circles respectively.
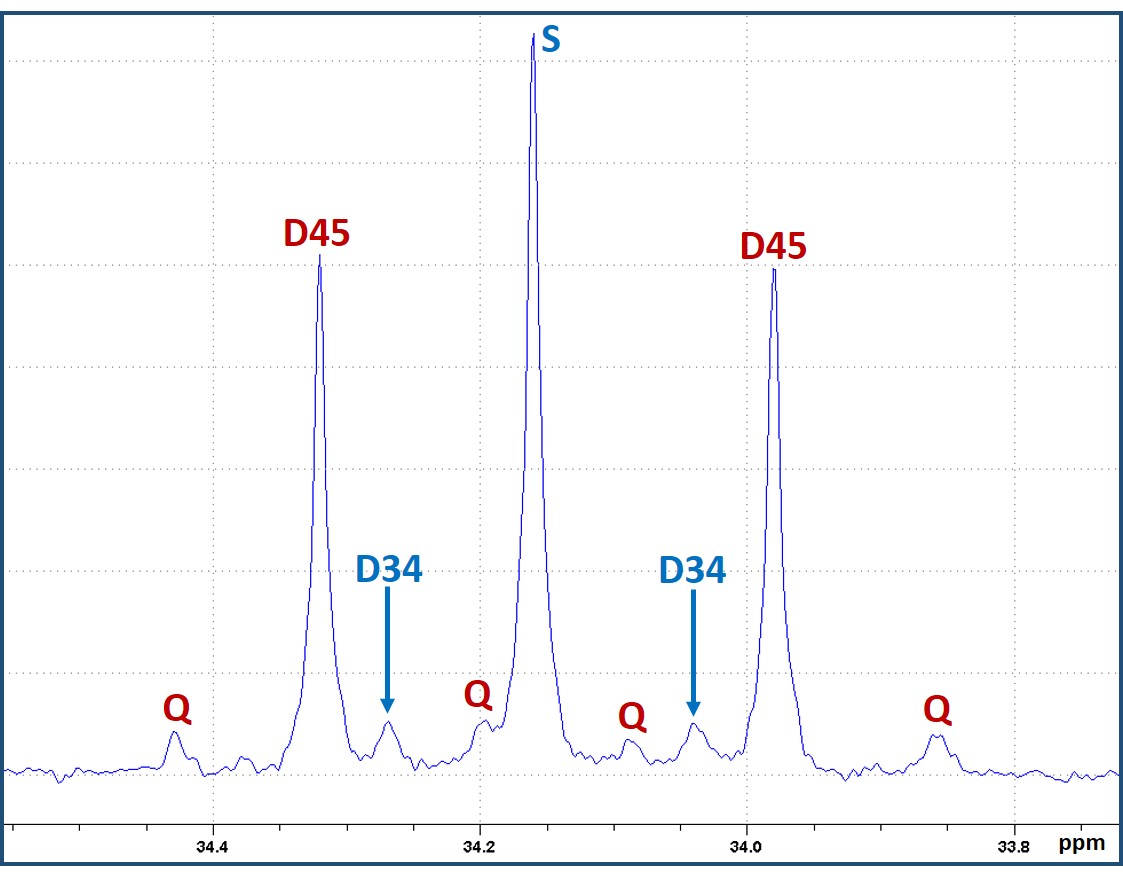
13C NMR spectral region of C4-glutamate signal with various spin-spin
coupled multiplets. The singlet (S) and the doublet (D34) are due to [4-13C]glutamate
and [3,4-13C]glutamate arising from the metabolism of [2,4-13C]BHB;
the doublet (D45) and the quartet (Q) are due to [4,5-13C]glutamate
and [3,4,5-13C]glutamate respectively that are derived from [U-13C]glucose.
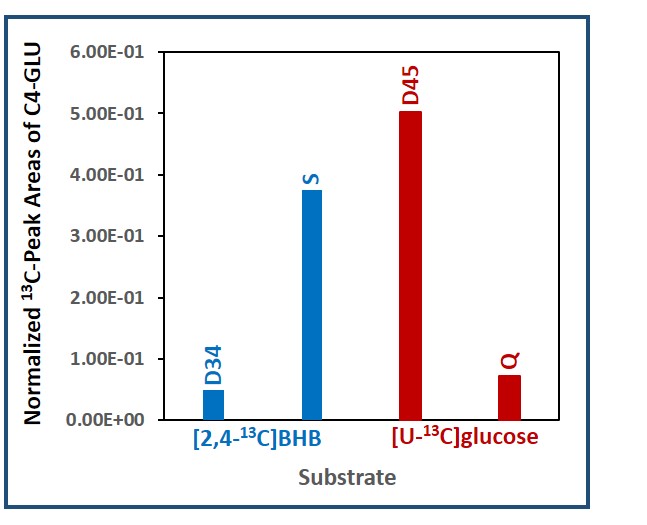
Normalized 13C NMR peak areas of various spin-spin coupled
multiplets of C4-glutamate. The singlet
(S) and the doublet (D34) are due to [4-13C]glutamate and [3,4-13C]glutamate
respectively that are derived from [2,4-13C]BHB. The doublet (D45)
and the quartet (Q) are due to [4,5-13C]glutamate and [3,4,5-13C]glutamate
respectively that are originating from [U-13C]glucose.