0336
Strategies for Obtaining Relaxation Rates due to Chemical Exchange from Parameter Free Atomistic Simulations1University of Oulu, Oulu, Finland, 2Oulu University Hospital, Oulu, Finland
Synopsis
Chemical exchange has likely a significant impact on the dispersion of T1ρ in cartilaginous tissue. In order to quantify this effect, we are developing atomistic simulation strategies for direct modelling of proton transfer reactions between biomacromolecules – first and foremost chondroitin sulfate – and water. Here we present benchmarking results from a number of semi-empirical quantum mechanics methods for several key system properties. Results suggest feasibility of the chosen approach.
Introduction
Chemical exchange of protons between the tissue matrix and water is a process with significance for several application areas of MRI. Most obviously for CEST, but most likely also for relaxation processes in the rotating frame of reference.1 We are most centrally interested in understanding the effect of different relaxation processes on the dispersion of T1ρ relaxation rates with varying spin-lock field. In order to quantify the impact of chemical exchange on T1ρ relaxation in cartilaginous tissues we are developing simulation strategies that can reliably represent the proton transfer processes between chondroitin sulfate – the component in cartilage that likely is the most important contributor to chemical exchange – and water. On one hand, the simulation of reactive processes, such as proton transfers, requires the explicit inclusion of electronic effects, making classical force-field methods inapplicable. On the other hand, the size of model system required to realistically represent any part of the tissue matrix is necessarily so large, that full quantum chemical treatment would be forbiddingly time-consuming even on the most modern supercomputers. We have therefore chosen to benchmark a number of semi-empirical methods, in which some of the quantum chemical terms are neglected or parametrised, for their capabilities to realistically represent proton transfer processes between chondroitin sulfate and water.Methods
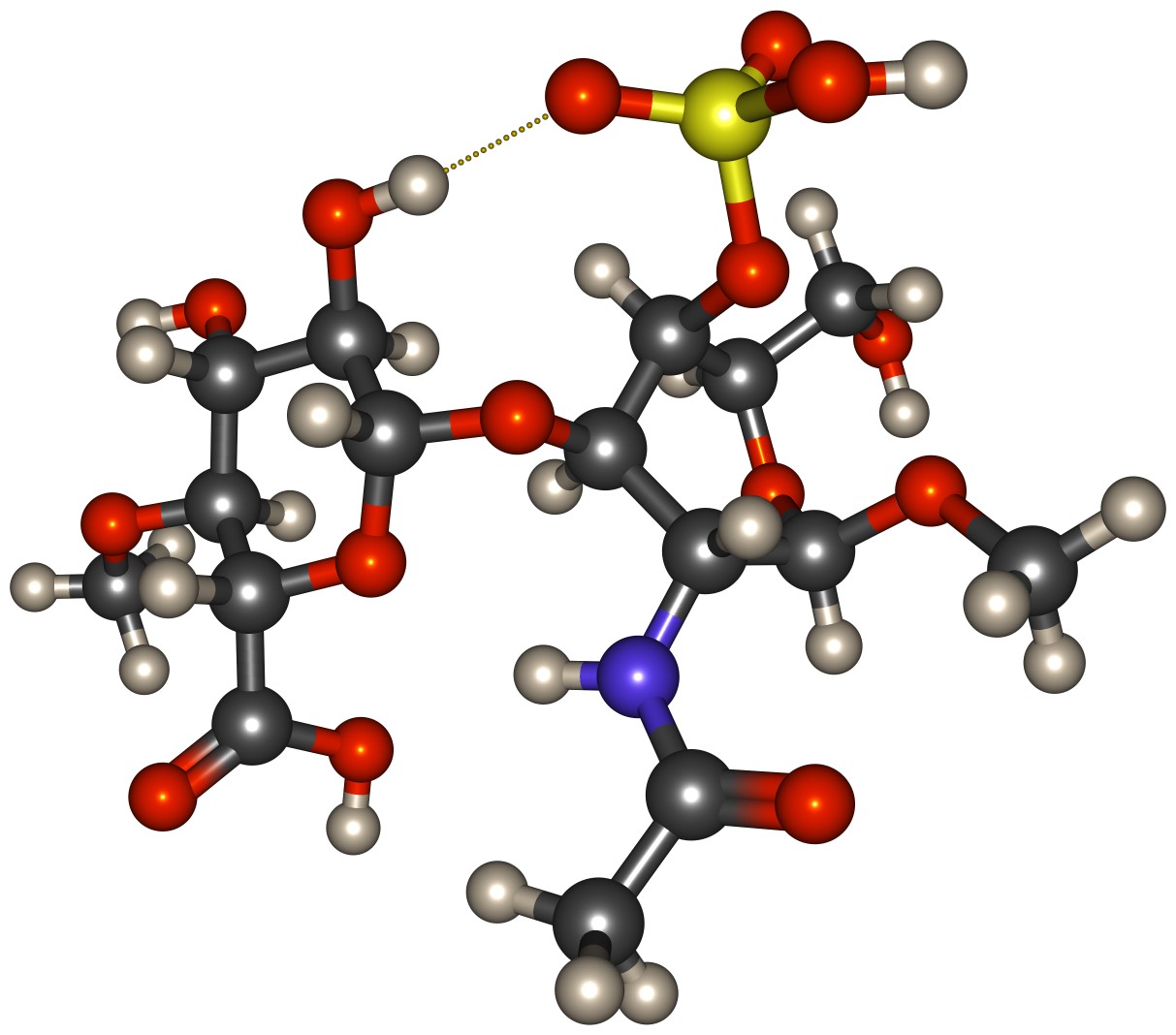
To determine the suitability of a number of different semi-empirical methods for modelling proton transfer reactions between chondroitin sulfate and water, we have undertaken a series of benchmark calculations. These calculations concern the ability of the methods to realistically describe (1) the structure of chondroitin sulfate, (2) the proton transfer from chondroitin sulfate to water, and (3) the solvation of protons in water. Our model system for chondroitin sulfate consists of a single disaccharide unit that is repeated in chondroitin-4-sulfate (β-GlcA-(1→3)-β-GalNAc4S), which is capped with methyl groups on both ends (cf. Fig. 1). In total, nine different semi-empirical methods (AM1n, DFTB3-D3, GFN-xTB, PM3n, PM6, PM6-DH+, PM6-D3H4, PM7, and RM1) were tested. Results obtained using the semi-empirical methods for the structure of the chondroitin sulfate fragment are compared to corresponding results on one hand from higher level quantum chemistry (density functional theory (DFT), ωB97XD/6-311++g(d,p)), and on the other hand from the carbohydrate-specific GLYCAM force field. Proton affinities of the chondroitin sulfate fragment were calculated step-wise for the different types of deprotonable functional groups: the bisulfate, and carboxylic groups were alternatively deprotonated in the first two steps, and hydroxyl groups, and the amide group were deprotonated as third step. The suitability of the semi-empirical methods for the dynamics of protons in water by calculations of the 2-D potential energy surface (PES) of the smallest structure realistically representing a hydrated proton, the Zundel ion (H5O2+), and the dynamics of proton transfers in isolated water clusters containing one excess proton. Both these characteristics are commensurate to the simulations performed by Wu et al. in their model reparametrisation study, which thus can also can be exploited as reference for the results.2Results & Discussion
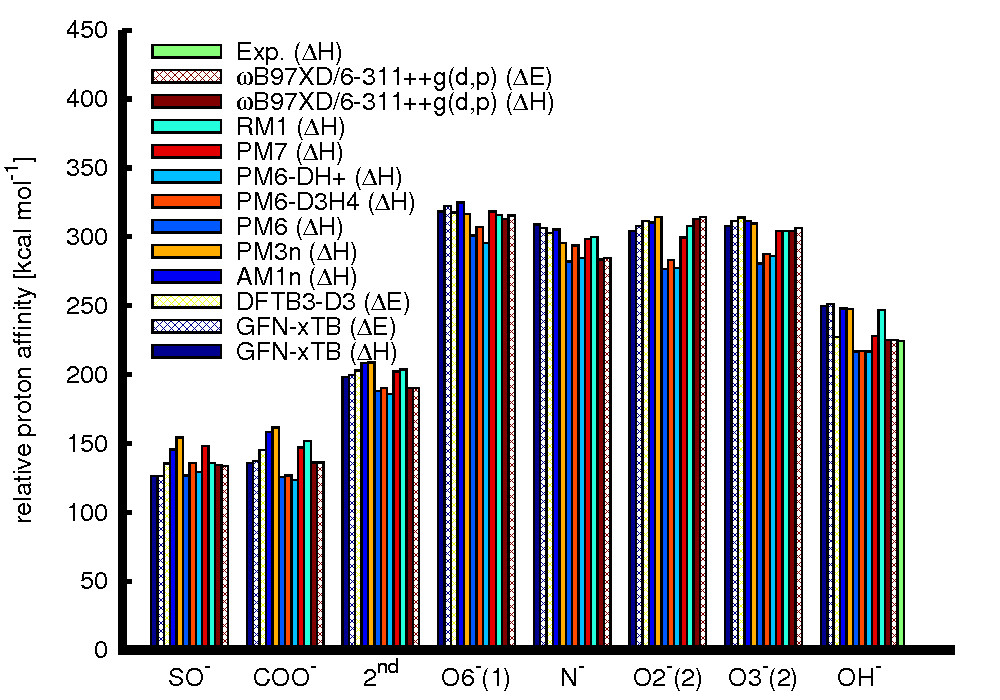
The results obtained for the gas phase proton affinities (i.e. the protonation enthalpy) of the various functional groups of the chondroitin sulfate fragment, and the hydroxide ion are summarized in Fig. 2. It should be noted here that different semi-empirical methods in part provide different measures – while most of the methods immediately only provide the enthalpy, the DFTB3-D3 method only provides the electronic energy. GFN-xTB, as well as the reference DFT method ωB97XD/6-311++g(d,p), provides both measures. The comparison shows comparatively poor performance of the AM1n and PM3n methods for the proton affinities of the acidic groups of the model fragment, and also of the PM6 based methods, especially for the hydroxyl and amide groups. Amongst the other methods, the best results are obtained from DFTB3-D3 in terms of both the mean absolute error, and root mean square error, and PM7 in terms of the mean signed error. Similarly, calculations on the Zundel ion show good qualitative agreement of most semi-empirical methods with the reference PES, with significant deviations only for the AM1n, and PM6 based methods. Simulations addressing the proton dynamics in water are still ongoing. Initial results indicate, however, that at least the PM7 method is capable of providing proton transfer rates that are comparable to estimates based on experiment.Conclusions
Our initial results support the hypothesis that realistic modelling of proton transfer processes between chondroitin sulfate model compounds and water is possible using a semi-empirical quantum mechanics approach. Further studies detailing the best method for the simulations in question are, however, still necessary.Acknowledgements
We thank the Academy of Finland (project 297033) for funding, and CSC-IT Center for Science in Espoo, Finland, for computing time.References
1. Chen E-L, Kim JK Magnetic Resonance Water Proton Relaxation in Protein Solutions and Tissue: T1ρ Dispersion Characterization. PLoS ONE 2010; 5(1), e8565.
2. Wu X, Thiel W, Pezeshki S et al. Specific Reaction Path Hamiltonian for Proton Transfer in Water: Reparameterized Semiempirical Models. J. Chem. Theory Comput. 2013; 9, 2672-2686.
Figures