0039
Fast and accurate white matter bundle segmentation1Division of Medical Image Computing, German Cancer Research Center (DKFZ), Heidelberg, Germany, 2Medical Faculty, University of Heidelberg, Heidelberg, Germany
Synopsis
Automatic white matter fiber bundle segmentation in diffusion-weighted MRI brain scans enables detailed studies of white matter characteristics in healthy and diseased brains. Existing approaches combine processing steps such as tractography, atlas registration and cortical parcellation, resulting in pipelines that are computationally intensive and tedious to set up. We present a novel convolutional neural network-based approach that incorporates or circumvents most of the usually required processing steps (no registration, no tracking, no parcellation). We demonstrate in 105 subjects from the Human Connectome Project that the proposed approach is much faster than existing methods while providing more accurate results.
Introduction
Automatic methods for the delineation of specific white matter fiber bundles from diffusion-weighted MRI (dMRI) brain scans enable in-depth analyses of white matter characteristics in healthy and diseased brains. Currently, existing approaches1-4 consist of complex pipelines that include several processing steps such as cortical parcellation, fiber tractography and registration. Such pipelines are computationally intensive and tedious to set up. We present a novel approach to direct white matter bundle segmentation that runs much faster than other methods and incorporates or circumvents most of the usually required processing steps. The setup is very convenient to use and at the same time provides the highest accuracy in a comparison to multiple benchmark methods on 105 subjects from the Human Connectome Project5 (HCP).Methods
We directly segment white matter bundles in dMRI images by employing a 2D fully convolutional neural network similar to the U-Net6. The network receives the three most dominant peaks of the fiber orientation distribution as its voxel-wise input. The peaks are encoded in a nine-channel input image after computing them using Constrained Spherical Deconvolution7. The network produces an individual binary segmentation channel for each bundle, thus segmenting all bundles included in the training process in one forward pass. Figure 1 provides an overview of the entire pipeline.
We used 105 HCP subjects to train and validate our model. Reference segmentations of 74 tracts per subject were created using a semi-automatic approach: First we created a whole brain tractogram with 10 million fibers per subject using probabilistic tractography8. Using TractQuerier2, we then extracted a rather coarse initial estimate of each bundle. To reduce the large number of false positive streamlines, we further filtered these tracts based on a combination of density and geometry constraints as well as automatic clustering9 and ROI-based filtering10 that required manual interaction. We additionally tested our model on data more typical for a clinical setting by downsampling the images to 2.5mm isotropic resolution and removing all but 32 weighted volumes at b=1000 s/mm2.
We compared our method with five state-of-the-art pipelines for white matter tract segmentation: TRACULA1, TractQuerier2, WhiteMatterAnalysis3 (WMA), RecoBundles4 and Diffeomorphic registration11 to a white matter bundle atlas that was created using the reference tracts (Atlas).
Results
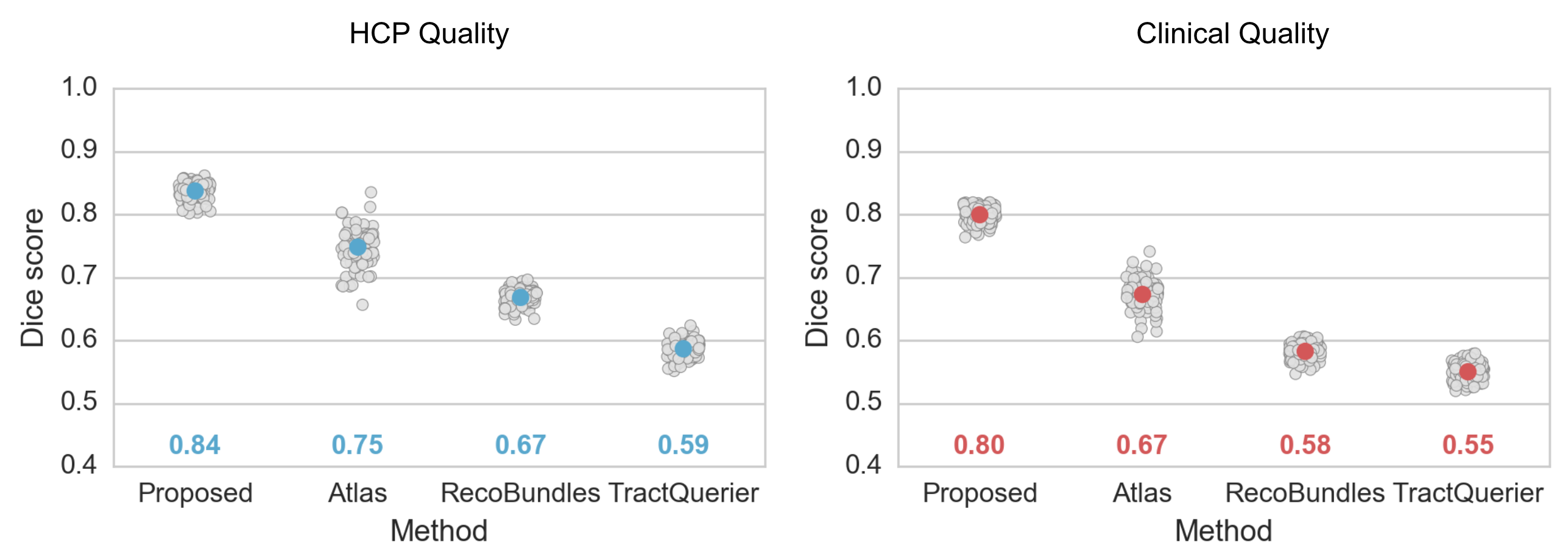
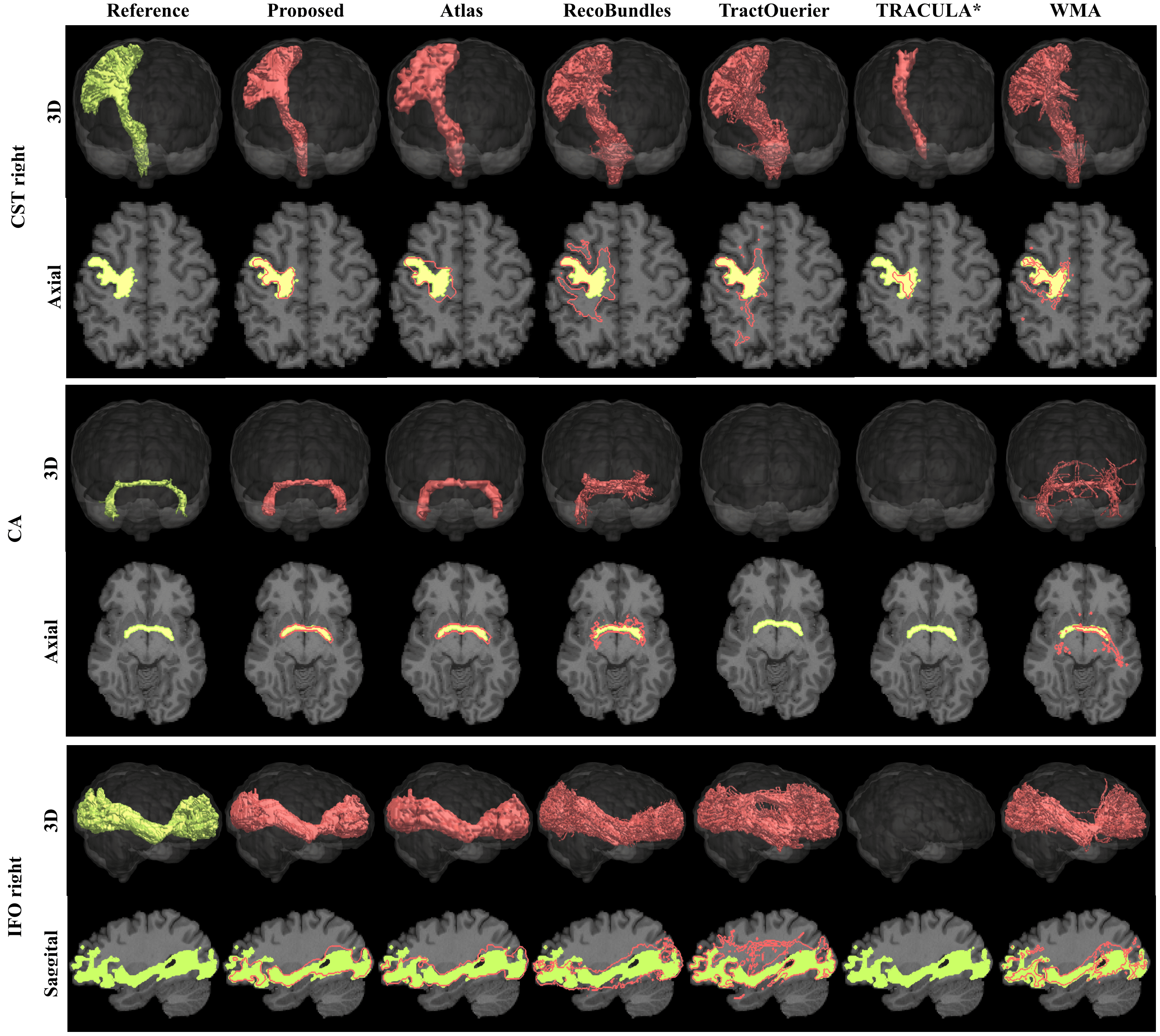
Figure 2 shows the Dice metrics obtained with all methods. The proposed approach significantly (p<0.001) outperformed Atlas, TractQuerier and RecoBundles by on average 17 points for HCP quality and 20 points for clinical quality data. We only qualitatively compared to TRACULA and WMA, as those two libraries use different reference tracts, making quantitative comparison difficult.Figure 3 gives a qualitative comparison of all methods on clinical quality data. Our approach yielded convincing results on all examined bundles.
Figure 4 compares the runtimes of all methods on a server with 48 2GHz Intel Xeon cores or an NVIDIA Titan X for the GPU-based approaches (TRACULA and the proposed method) respectively. On the clinical quality data our method is 460x faster than the average runtime of all other methods, on the HCP quality data it is 50x faster.
Discussion
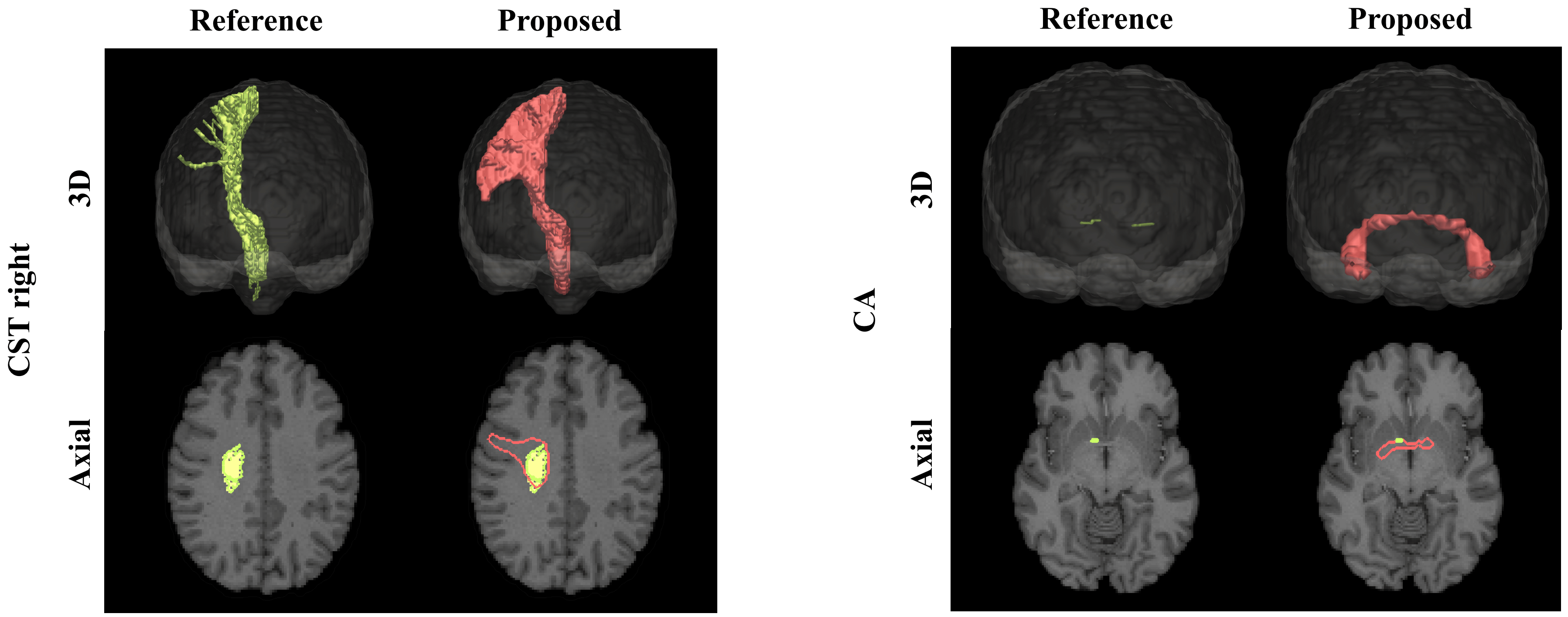
The results demonstrate a strong potential of the proposed direct bundle segmentation method, especially when considering difficult tracts like the CST and the CA. Given the margin by which the reference methods were outperformed, the results are highly encouraging to further pursue this direction of research. One limitation of supervised techniques like the proposed one is obvious though: they always depend on the availability and quality of training data. Interestingly, the training data in some subjects missed some of the difficult parts of the CA or the lateral projects of the CST, since the probabilistic tractography did not successfully reconstruct these parts. A post-hoc direct comparison of our method’s results with these reference tracts revealed that our method was able to yield more complete reconstructions than the reference method (figure 5). A further limitation of the presented work is that we so far only tested our method on healthy subjects. In the future we will extend it to pathological cases.Conclusion
The proposed method segments 74 different major white matter bundles with consistently high accuracy even on low resolution data. It takes only 1.7 minutes per subject to segment all bundles and no further tools for registration or tracking are needed. Although our method was only trained on HCP data, it also shows promising results on other non-HCP datasets due to the data augmentation used during training. The code of our method is available as an easy to use python package: https://github.com/MIC-DKFZ/TractSegAcknowledgements
This work was supported by the German Research Foundation (DFG) grant numbers MA 6340/10-1 and MA6340/12-1.References
1. Yendiki et al. Automated probabilistic reconstruction of white-matter pathways in health and disease using an atlas of the underlying anatomy. Front. Neuroinform. 5(23), 12–23 (2011)
2. Wassermann et al. The white matter query language: a novel approach for describing human white matter anatomy. Brain Struct. Funct. 221(9), 4705–4721 (2016)
3. O'Donnell et al. Automatic tractography segmentation using a high-dimensional white matter atlas. Medical Imaging, IEEE Transactions on 26.11 (2007): 1562-1575.
4. Garyfallidis et al. Recognition of white matter bundles using local and global streamline-based registration and clustering. NeuroImage (2017)
5. Glasser et al. The minimal preprocessing pipelines for the Human Connectome Project. NeuroImage (2013)
6. Ronneberger et al.: U-net: Convolutional networks for biomedical image segmentation. In: MICCAI. pp. 234–241. Springer (2015)
7. Jeurissen et al. Multi-tissue constrained spherical deconvolution for improved analysis of multi-shell diffusion MRI data. NeuroImage (2014)
8. Tournier et al. Improved probabilistic streamlines tractography by 2nd order integration over fibre orientation distributions. In: Proc. ISMRM. p. 1670 (2010)
9. Garyfallidis et al. Quickbundles, a method for tractography simplification. Frontiers in Neuroscience (2012)
10. Stieltjes et al.: Diffusion Tensor Imaging - Introduction and Atlas. Springer Berlin Heidelberg (2013)
11. Avants et al. Symmetric Diffeomorphic Image Registration with Cross-Correlation: Evaluating Automated Labeling of Elderly and Neurodegenerative Brain, 12(1), 26-41 (2009)
Figures