5393
How does chronic neuroinflammation affect resting state functional connectivity?1Neurology, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States, 2Biomedical Research Imaging Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States, 3Neurobiology Laboratory, National Institute of Environmental Health Sciences, RTP, NC, United States, 4University of North Carolina at Chapel Hill, Chapel Hill, NC, United States, 5Biomedical Research Imaging Center, University of North Carolina at Chapel Hill, 6Neurobiology Laboratory, National Institute of Environmental Health Sciences, 7National Institute of Environmental Health Sciences, RTP, NC, United States
Synopsis
Chronic neuroinflammation, present in most neuropathologies, has long-term consequences on neurocircuit connectivity synchrony and strength. By implementing multi-modal techniques to quantify neuroinflammation, we found a strong association linking the intensity of neuroinflammation with depressed functional connectivity. We partly attribute these changes to neurodegeneration and the loss of central NE.
Purpose
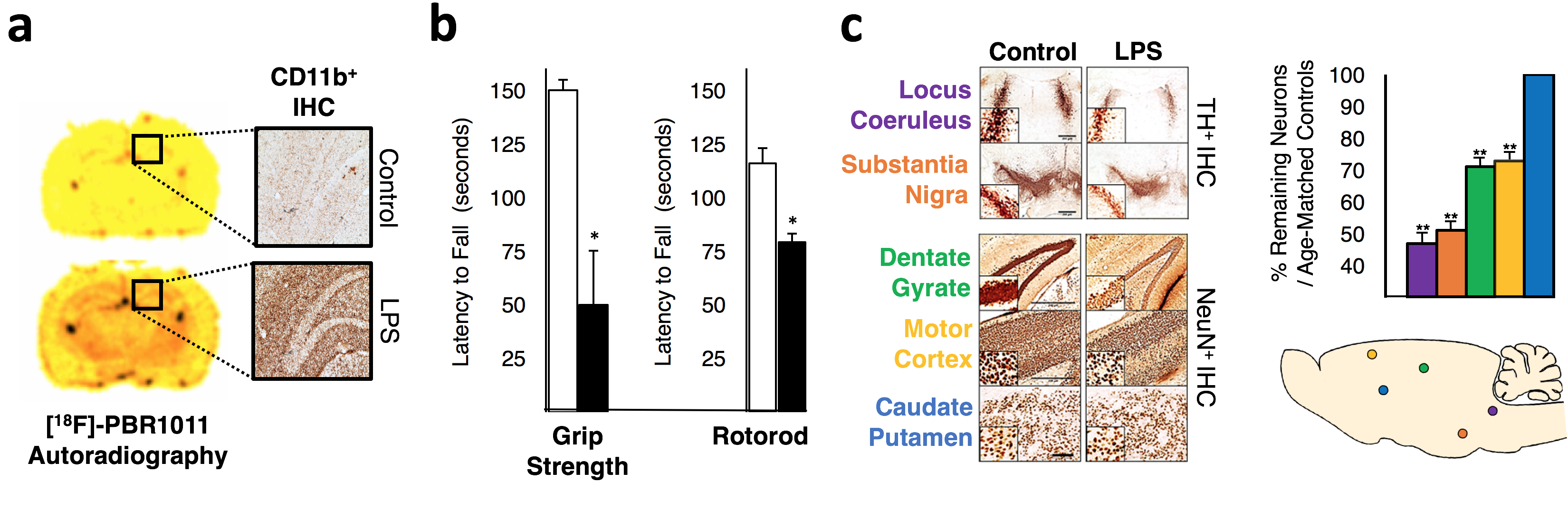
Neuroinflammation serves as a defensive response to isolate, repair or destroy injurious brain tissue that has been damaged by pathogens, senile plaques, blunt trauma, ischemia or oxidant stress.1 When neuroinflammation is persistent, such as in Alzheimer's disease, Parkinson's disease, Huntington's disease, and Multiple Sclerosis, it contributes to the degeneration of susceptible bystander neurons from microglia-mediated collateral oxidant damage.2 Though non-immunologically activated microglia are involved in plasticity to improve functional connectivity3, almost nothing is known about how their immunological activation during chronic neuroinflammation alters neurocircuit functions. Most importantly, deciphering how chronic neuroinflammation alters fcMRI is clinically relevant for early diagnosis of neurodegenerative disorders since patients present chronic neuroinflammation nearly a decade before the onset of their disorder-related symptoms. To investigate this, mice were systemically exposed to the bacterial endotoxin lipopolysaccharide (LPS) to generate a global, chronic neuroinflammation4,5 detected 12 months after onset using the inflammatory markers [18F]-PBR111 and CD11b+ (Fig 1a). This model afflicts motor balance and coordination (Fig 1b) and recapitulates the progressive pathological axis of many neurodegenerative disorders with high neuronal losses of noradrenergic Locus Coeruleus (NE-LC) and dopaminergic Substantia Nigra pars compacta (DA-SNpc) neurons and lesser losses to hippocampal and motor cortex neurons while sparing the striatum and VTA (Fig 1c). Since heterogeneous intensities in neuroinflammation are present in this model and they seem to align with regions of degeneration, we hypothesize that chronic neuroinflammation affects functional neurocircuit synchrony and strength in an intensity-depended manner. Using a multi-modal approach combining in vivo PET and fcMRI we interrogated how spatial gradations in chronic neuroinflammation intensities throughout the brain directly alter the strength of functional connectivity synchrony and neuronal metabolism in resting state neural circuits.Methods
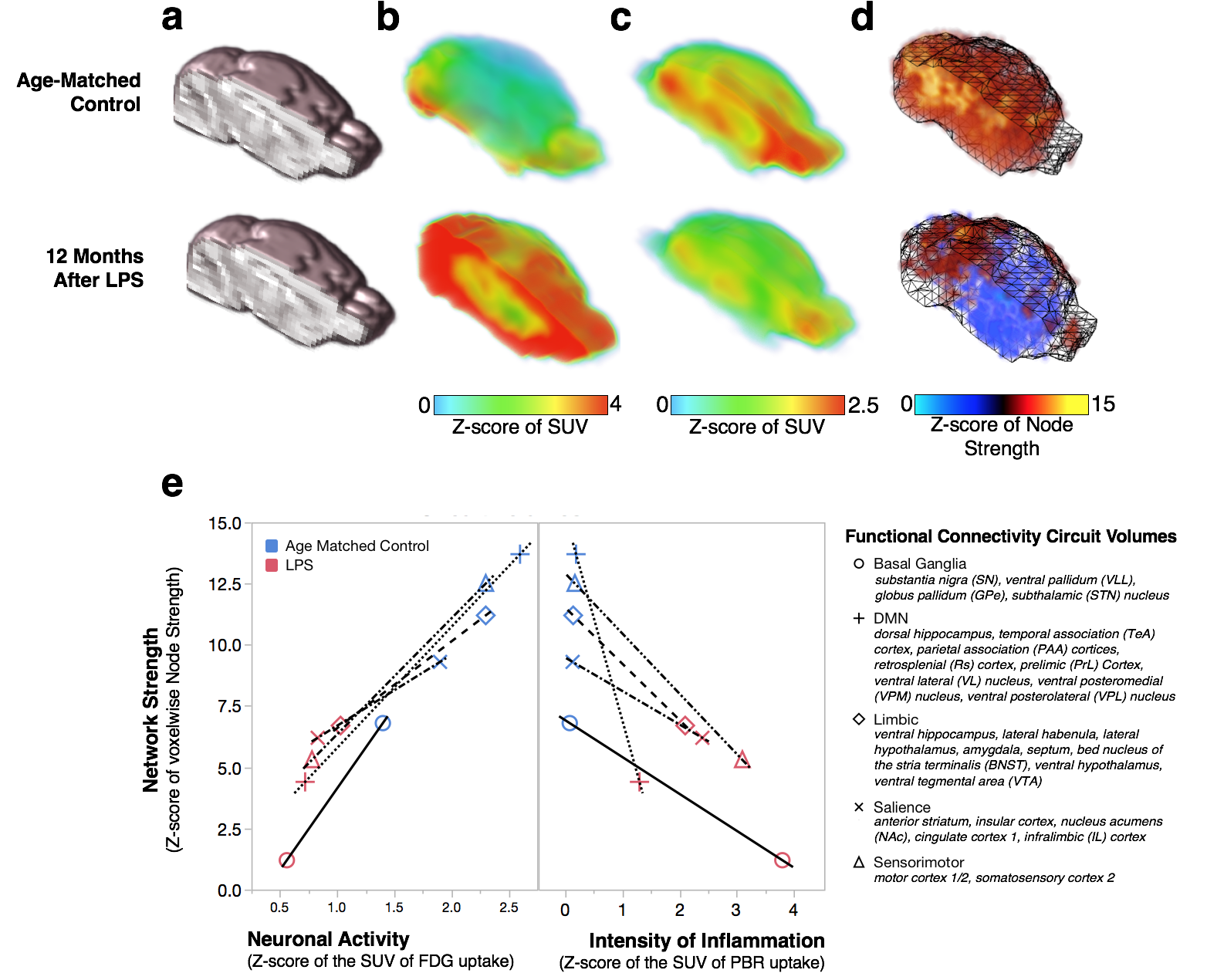
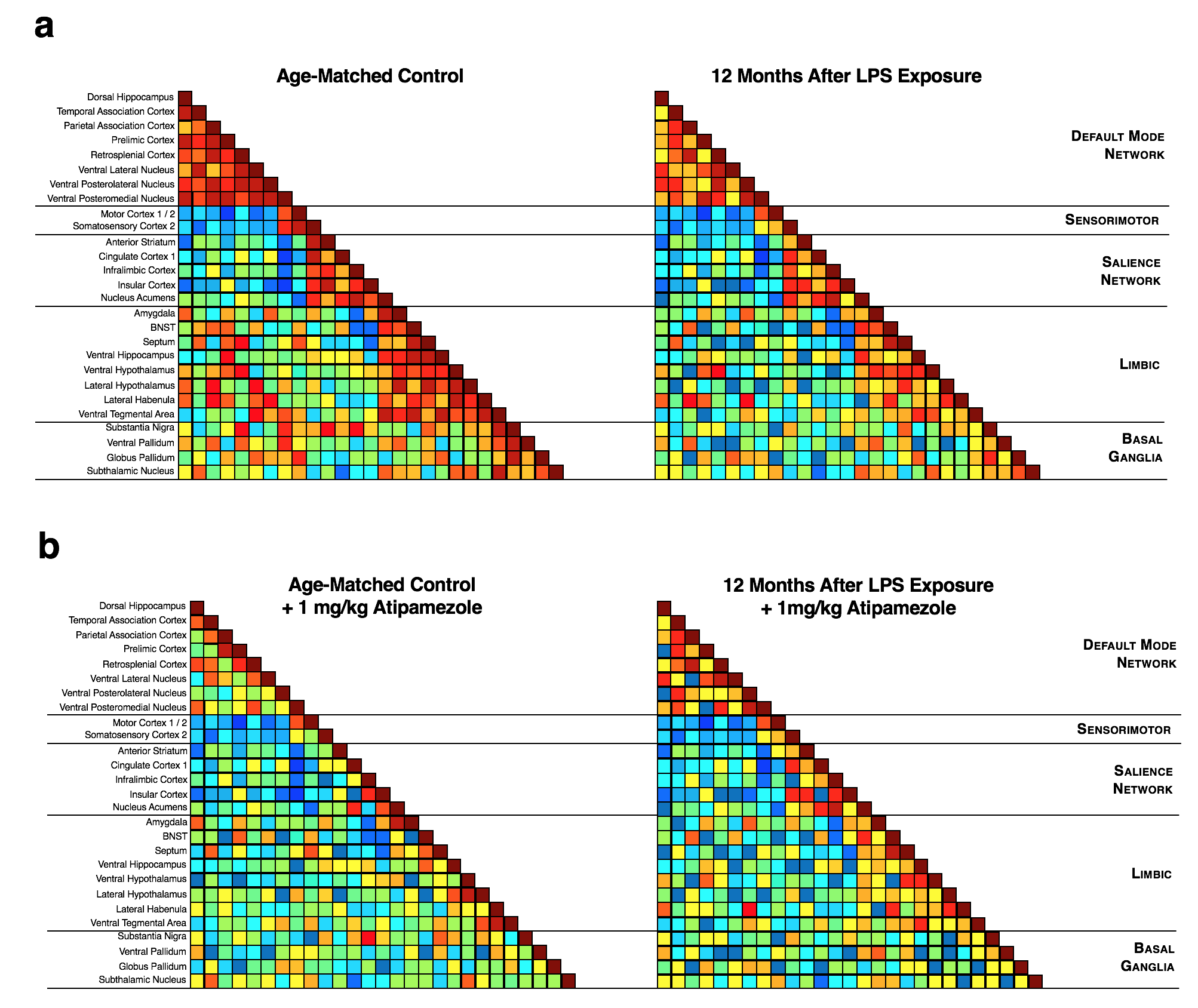
C57BL6/J mice were exposed to saline (n=12) or 5 mg/kg of LPS (i.p, E. coli strain O111:B4, Sigma-Aldrich L3012; n=12). Global chronic neuroinflammation was confirmed in vivo on a GE eXplore Vista PET/CT using by [18F]-PBR1116 and [18F]-FDG-PET was also performed as a brain activation surrogate. PET radiotracers (~10 MBq, i.v.) were administered 45 min prior to imaging and mice were scanned under 2% isoflurane for a 20 min PET static emission acquisition correcting for decay, attenuation, scatter and random coincidences and a 6 min CT acquisition for skull stripping. Brain images were aligned to custom templates and standardized uptake values (MBq/cm3) were calculated and transformed into normally distributed Z-scores for each function connectivity circuit volume (listed on Fig 2e) in PMOD. fcMRI were collected on a Bruker 9.4-Tesla/30-cm scanner with a BGA9-S gradient insert and imaged using a Bruker mouse receiver surface coil coupled with a transmitter volume coil. Imaging sedation of 1-1.5% isoflurane and subject physiological parameters were maintained while acquiring single-shot gradient echo EPIs in two, 10 minute sessions with bandwidth = 250 kHz, TR/TE = 3000/10 ms, labeling duration = 3.0 s with no post-labeling delay, matrix = 64x64, FOV = 1.92 cm2, slice number = 26 and slice thickness = 0.3 mm. BOLD acquisitions were slice-timing and motion corrected, aligned, spatial smoothed (FWHM = 0.6 mm), bandpass filtered (0.01-0.1 Hz) and normalized using FSL and warped with ANTs to a custom C57BL/6J probabilistic EPI atlas. Correlation maps were generated using Pearson correlations undergoing a Fisher's r-to-Z transformation through JMP (v.13, SAS) and visualized as connectivity matrices. Voxelwise functional connectivity maps were generated using AFNI and Z-scores for circuit strengths were calculated for each function connectivity circuit volume and compared to PET data.Results
Brain regions with greater chronic neuroinflammation intensities (Fig 2b) had suppressed neuronal metabolism as measured by PET-FDG (Fig 2c, e), reduced voxelwise connectivity strengths in function connectivity circuit volumes (Fig 2d, e) and decreased resting-state circuit synchrony (Fig 3a). Notably, chronic neuroinflammation significantly reduced connectivity strength (p<0.05) in all five major resting-state network compared to controls. Previous findings7 lead us to interrogate whether NE loss from LC degeneration could alter fcMRI connectivity during chronic neuroinflammation by exposing mice to atipamezole (Fig 3b, 1 mg/kg, s.c.) to evoke fcMRI changes through a 30% increase in central NE levels8 and found mice with chronic neuroinflammation had poor responses in fcMRI networks synchrony compared to age-matched controls (Fig 3).
Discussion
Though inflammation can suppress functional connectivity in the hedonic reward circuits of patients with depression9, our study is the first to directly correlate depressions in both synchrony and circuit strengths in all major resting-state networks directly to gradations in the intensity of chronic neuroinflammation intensity. Furthermore, we suspect these changes to be a result of significant neurodegeneration and the loss of cortical NE.Acknowledgements
We thank members of the Shih and Hong laboratories for their valuable input discussing the experimental design and results in this study. We also thank Jon Frank and Joseph Merrill of the University of North Carolina at Chapel Hill’s Biomedical Research Imaging Core for performing the PET imaging studies. E.A.O. was supported by the National Heart, Lung, and Blood Institute (NHLBI) Training Grant. M.D. was supported by the Human Frontier Science Program-Cross Disciplinary Fellowship. S.S. and J.S.H. were supported my Intramural Research Division of the National Institute of Environmental Health Sciences (NIEHS). Y.Y.I.S. was supported by the NINDS (NS091236), the National Institute of Mental Health (MH106939), the National Institute on Alcohol Abuse and Alcoholism (AA020023), the National Institute of Health UL1TR001111 sub-awards 550KR81420 and 550KR91413, the Brain and Behavior Foundation Young Investigator Award and Ellen Schapiro & Gerald Axelbaum Investigator fund, the American Heart Association Scientist Development Award (15SDG23260025), and the Department of Neurology and the Biomedical Research Imaging Center at UNC Chapel Hill.References
1. Saijo K GC. Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol. 2011;11:775-787
2. Block ML ZL, Hong JS. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57-69
3. Zhang J, Malik A, Choi HB, Ko RW, Dissing-Olesen L, Macvicar BA. Microglial cr3 activation triggers long-term synaptic depression in the hippocampus via nadph oxidase. Neuron. 2014;82:195-207
4. Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, et al. Systemic lps causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453-462
5. Wang Q, Qian L, Chen SH, Chu CH, Wilson B, Oyarzabal E, et al. Post-treatment with an ultra-low dose of nadph oxidase inhibitor diphenyleneiodonium attenuates disease progression in multiple parkinson's disease models. Brain. 2015;138:1247-1262
6. Van Camp N, Boisgard R, Kuhnast B, Theze B, Viel T, Gregoire MC, et al. In vivo imaging of neuroinflammation: A comparative study between [(18)f]pbr111, [ (11)c]clinme and [ (11)c]pk11195 in an acute rodent model. Eur J Nucl Med Mol Imaging. 2010;37:962-972
7. Oyarzabal EA DM, Lee SH, Sciolino N, Evsyukova I, Jensen P, Shih YY. Deciphering the functional role of locus coeruleus-derived norepinephrine using chemogenetic fmri and 18fdg-pet. 24th Annual ISMRM Resarch Meeting. 2016
8. Bekar LK, Wei HS, Nedergaard M. The locus coeruleus-norepinephrine network optimizes coupling of cerebral blood volume with oxygen demand. J Cereb Blood Flow Metab. 2012;32:2135-2145
9. Felger JC, Li Z, Haroon E, Woolwine BJ, Jung MY, Hu X, et al. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol Psychiatry. 2015
Figures