4885
Cardiac Magnetic Resonance for Characterizing a Spontaneous Hypertrophic Cardiomyopathy Mouse ModelMin-Chi Ku1, Till Huelnhagen1, Andreas Pohlmann1, and Thoralf Niendorf1,2,3
1Berlin Ultrahigh Field Facility, Max Delbrück Center for Molecular Medicine in the Helmholtz Association, Berlin, Germany, 2Experimental and Clinical Research Center, Charite Medical Faculty and the Max Delbrueck Center for Molecular Medicine in the Helmholtz Association, Berlin, Germany, 3DZHK (German Centre for Cardiovascular Research), partner site Berlin, Germany
Synopsis
HCM is the most common inherited heart disease. The two most frequently seen mutated genes are MYH7 and MYBPC3 which account for nearly 80% of familiar HCM. In this study we hypothesized that these gene variants will affect both LV and RV function. By in-vivo CMR we detected LV hypertrophy in a mouse strain DBA/2J bearing the two gene variants. Interestingly there is no defected LV function found but changes in RV function as both male and female DBA/2J mice had declined RVEF. Our results provide new insights into the correlation of genetic alteration and HCM phenotype.
Introduction
Hypertrophic cardiomyopathy (HCM) is the most frequent genetic heart disease, a leading cause of sudden death in young adult, and the cause of heart failure symptoms at any age. HCM is defined as thickening of left ventricular (LV) wall without any other cardiac or systemic condition. There are at least 20 genes encoding sarcomere or sarcomere-related proteins which have been heavily studied in HCM. Among those genes, the two most frequently seen mutated genes are MYH7 (encoding myosin heavy chain-beta) and MYBPC3 (encoding myosin-binding protein C) which account for nearly 80% of familiar HCM. Since cardiac function is not compromised at early stages, affected individuals are often unaware of the disease until complications arise. CMR is a very well accepted tool for the characterization of diverse HCM phenotypes. However, HCM shows extremely heterogeneous phenotypes. Even in same families bearing the same genetic defects, the disease presents with various phenotypes and clinical symptoms indicating that the relationships between genotype and phenotype expression in HCM is complicated. Therefore, finding a predictable relation between genotype and disease expression is needed for clinical decision-making and for prognosis. Because of the availability of genetically engineered murine models, in-vivo high-resolution CMR of mice is of high relevance and value for studying HCM. It gives rise to valuable information which can be translated to the human findings. For example, HCM prevalently involves the LV, but in one third of cases it is biventricular. There is growing evidence that right ventricular (RV) dysfunction impacts disease progression. However, attempts to evaluate both LV and RV function correlates genetic defect in HCM are limited. Thus, the aim of our study was to examine the possible phenotypes associated with both genes MYH7 and MYBPC3. We used a mouse strain bearing both gene variants[1] and evaluated their LV and RV morphology and function by in-vivo CMR.Methods
Eight months old male (n=3), female C57BL/6J (n=4) mice as control group, and male (n=4), female DBA/2J (n=4) mice bearing variants on MYH7 and MYBPC3 were used for CMR analysis on a 9.4T animal MR scanner (Biospec 94/20, BrukerBiospin, Germany). All images were acquired using a cryogenic transceiver quadrature RF surface coil (CryoProbe, BrukerBiospin, Germany). To obtain a stack of cardiac short axis views covering the whole mouse heart, ten slices were consecutively acquired using self-gated bright-blood cine imaging (IntraGate-FLASH, TE/TR= 1.58/8.5ms, FA = 20°, BW = 98kHz, FOV=11×22mm2, matrix=192×384, slice thickness=0.8mm, cardiac frames=16)[2]. Cardiac function assessment was performed using CMR42 (Circle CVI, Canada) and analyzed on a slice-by-slice basis. Endo- and epi-cardiac borders were manually segmented in end-systole and diastole using a stack of short axis images. Ejection fractions (EF) and myocardial masses (diastole and systole) were calculated for left and right ventricle. After in-vivo MRI, mice were perfused with 4% PFA. Fixed ex-vivo hearts were imaged at high spatial resolution (RARE, TE/TR= 40.7/2200ms, FOV=15×10mm2, matrix=500×336, slice thickness=0.3mm, in-plane spatial resolution=30µm).Results
In FLASH-CINE images, both male and female DBA/2J mice showed LV wall thickening versus controls (Fig.1). The difference of LV wall thickness in female DBA/2J and C57BL/6J is less pronounced but is still significant (Fig.2). Both male and female DBA/2J mice also showed increased LV masses (P=0.017, male; P=0.0013, female). Although LV wall thickness is increased in DBA/2J mice, LVEF was similar to C57BL/6J mice (P=0.26, male; P=0.27, female). Interestingly, both male and female DBA/2J mice had declined RVEF (P=0.03, male; P=0.04, female). High-resolution ex-vivo images revealed subtle anatomical details and confirmed the LV wall thickening (Fig.3). A closer examination of the coronal view of the mouse heart obtained from male C57BL/6J and DBA/2J mice revealed no obvious myocardial crypts when using a spatial resolution as good as 30µm.Discussion and Conclusions
We evaluated the phenotypic impact of MYH7 and MYBPC3 sarcomere gene mutations on murine cardiac structure and function using CMR. We found hypertrophic LV in DBA/2J mice, as well as relevant changes in mouse RV function in these mice when compared to C57BL/6J mice. Previous studies revealed that RV involvements were comparable to those of LV global function impairments in HCM patients[3]. Thus, the presence of RV dysfunction on the MRI may help to predict the severe symptomatic HCM related heart abnormalities and to study the HCM progression and symptom occurrence. Furthermore, studying the phenotype on distinctive genetic mutation mouse models can provide insights into whether LV hypertrophy is the primary trait, or whether it occurs as a secondary adaptive response in HCM. In conclusion, our result showed changes in RV and provide new insights into the correlation of genetic alteration and HCM phenotype.Acknowledgements
No acknowledgement found.References
[1] Zhao, et al., PLoS One. 10: e0133132. (2015)
[2] Wagenhaus, et al., PLoS One. 7: e42383. (2012)
[3] Zhang et al., PLoS One. 9: e104312. (2014)
Figures
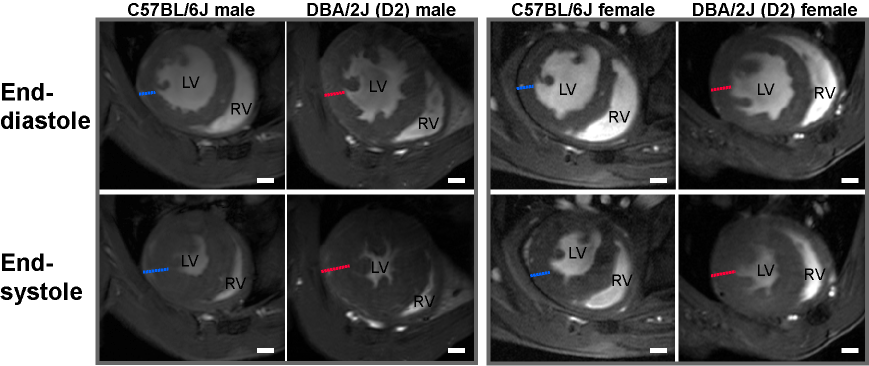
Figure 1: Mice bearing both gene variant on MYH7 and MYBPC3 show LV hypertrophy. Representative FLASH
CINE images (diastole and systole) showing a mid-ventricular short axis view in
both male and female C57BL6J and DBA/2J mouse heart at 9.4T. Blue dash lines depict the diameter of
myocardium in C57BL6J mice. Red dash lines depict the diameter of myocardium in
DBA/2J mice. Scale bar=1mm
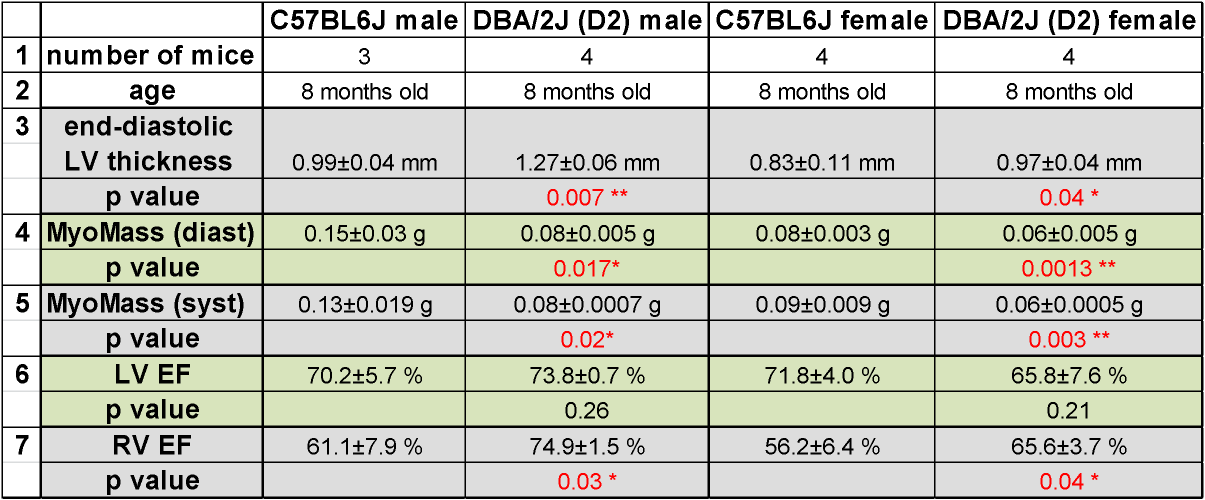
Figure 2: Mice bearing both gene variant on MYH7 and MYBPC3 show
different heart morphology and function. * p<0.05; **p<0.01
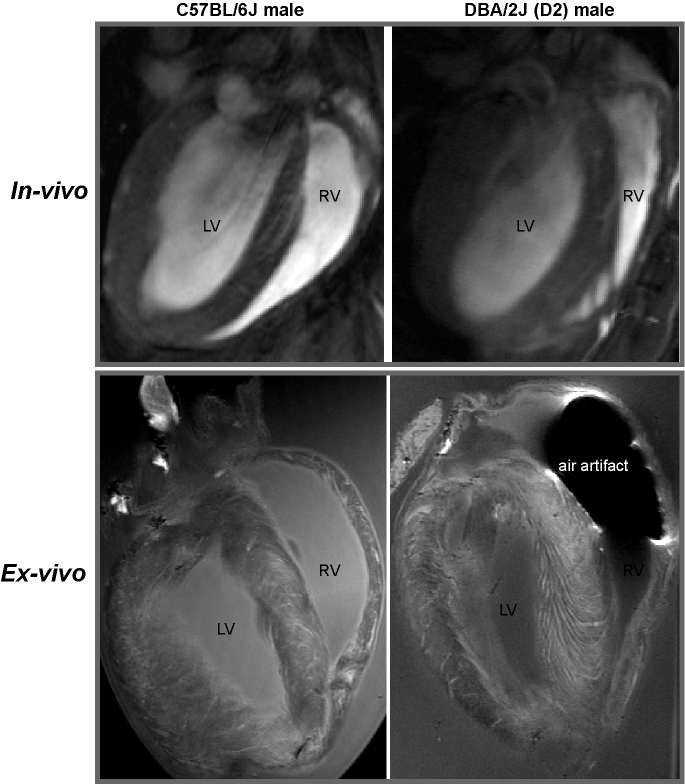
Figure 3: In-vivo and ex-vivo heart images of C57BL6J and DBA/2J mice. From both in-vivo and high resolution ex-vivo images of coronal view, no myocardial
crypts can be seen with the spatial resolution used.