4536
Chronically elevated Taurine in the putative seizure onset zone during posttraumatic epileptogenesis identifies epilepsy-prone rats1Neurobiology, A.I.Virtanen Institute, Univ. Eastern Finland, Kuopio, Finland
Synopsis
In lateral fluid percussion rat model for posttraumatic epilepsy 50% of animals have slowly developed epilepsy 1 year after the head injury.1 The seizures presumably originate in the perilesional cortex that appears normal in conventional MRI.2 We targeted localized magnetic resonance spectroscopy (MRS) to this cortical area 6 months post-injury, and found elevated Taurine and elevated macromolecule concentration to differentiate the subpopulation (19%) of injured animals with higher susceptibility to seizures in EEG recorded PTZ test. The preliminary immunohistochemical analysis of the underlying complex pathology revealed swollen neurons that may associate with the increase of osmoregulator taurine.
INTRODUCTION
Traumatic brain injury (TBI) launches complex cascades of progressive degenerative and regenerative processes, and may lead to epilepsy.1 Preclinical studies utilizing the lateral fluid percussion injury (LFPI) rat model for posttraumatic epilepsy have recently suggested that late onset seizures in posttraumatic brain originate from the cortex close to the initial impact injury site.2,3 Perilesional cortex, the surviving cortex surrounding the necrotic primary lesion, is known to undergo neovascularization and BBB permeability associated inflammatory response, different forms of gliosis, neurodegeneration, demyelination, changes of the extracellular matrix and shift in energy metabolism.4-6 However, it is still not understood, which of these changes have the key role in establishing the epileptic condition. We set out to study the neurochemical profile in the chronic stage 6 months after TBI with advanced pathophysiology. The study cohort did not yet have spontaneous seizures at this time. We correlated MRS with seizure susceptibility measures determined later by EEG. We hypothesized that non-invasive proton MRS targeted to the perilesional cortex can differentiate the epileptogenic and non-epileptogenic rats, and that a specific imbalance in neurochemical concentrations will serve as biomarker for posttraumatic epilepsy.METHODS
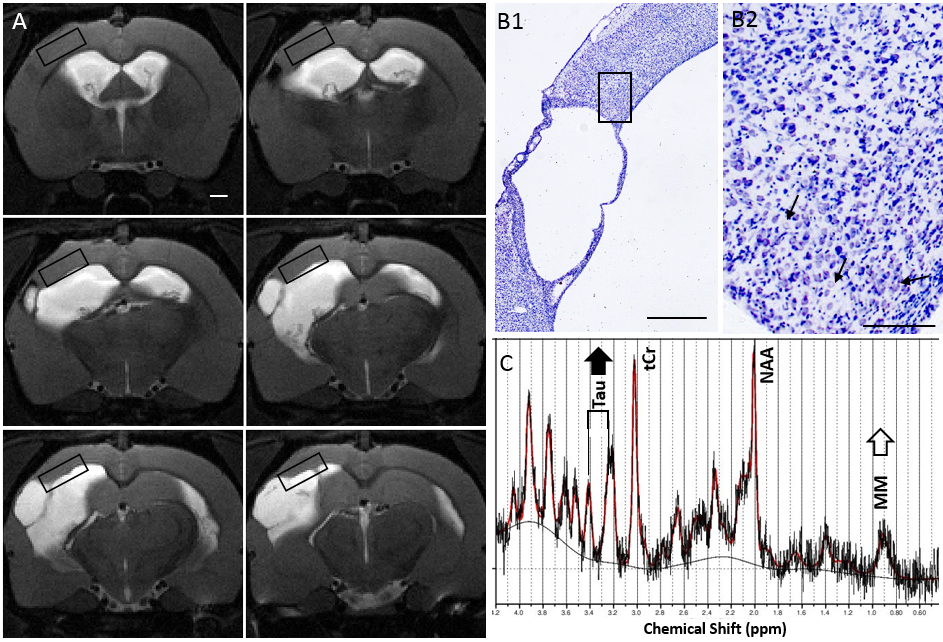
Adult male Spraque Dawley rats (22 TBIs, 10 shams) were induced with lateral fluid percussion injury. Six months after injury the neurochemical profile in the perilesional cortex was measured by localized 1H-MRS targeting normal-appearing cortical tissue adjacent in rostro-dorsal edge of the primary cystic lesion with hematomas. The lesion morphology and cortical thinning forced the voxel dimensions to 1x3x5mm (Fig.1A). Spectra were obtained at 9.4T by PRESS (TE 11ms, TR 2500ms, 320/640 averages for sham/TBI, MAPSHIM, VAPOR water suppression, and reference water) and analyzed by LCmodel including metabolites with mean SD%<20. [For taurine SD% 15.2 ± 5.2, for MM09 19.9±5.4, for MM09+Lip09: 16.4±3.3. No lipids were observed at 1.3ppm.] Absolute concentrations were corrected for cerebrospinal fluid (CSF) volume fraction and ratios Tau/total Creatine and Tau/NAA calculated. Seven months post-injury epileptiform activity was recorded in continuous 4wk video-EEG and pentylenetetrazol test (PTZ; 25mg/kg i.p., cortical screw electorodes). Latency to first spike after PTZ inj. was used as the measure for seizure susceptibility, and animals with latency <128sec (sham mean-1SD) were classified as epileptogenic. Statistics: Strongest bivariate Pearson correlations between neurochemicals and latency to spike were subjected to ROC analysis. Cellular cytoarchitecture was analyzed in Nissl stained brain sections.RESULTS and CONCLUSIONS
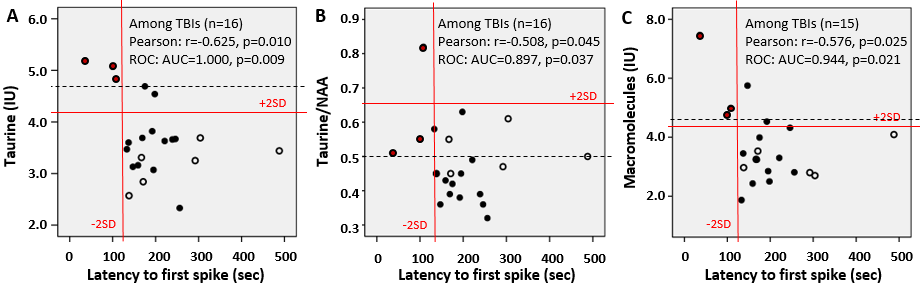
Six months after the head injury - among the several neurochemical abnormalities present - only the elevated taurine and elevated macromolecules were found to correlate with time to the first epileptiform spike after administration of PTZ, and identify the epileptogenic animals among the TBI cohort (Fig.2). The higher the taurine the sooner the spiking started after PTZ. Similar results were obtained by using absolute Tau, Tau /tCr, and Tau/NAA concentrations indicating that taurine elevation was independent of total cellular and neuronal count. Neither Tau nor MM correlated with CSF volume fraction indicating tissue origin. The area of interest is highly challenging for localized spectroscopy and the methodological approach was driven and restricted by the a priori knowledge of the brain areas affected (raw data in Fig1C). Immunohistochemical stainings are to be tailored to explain the high taurine, but the preliminary observations about cellular architecture in Nissl staining revealed swollen neurons in the close vicinity of the cystic lesion still 6mo after TBI (not across the MRS voxel, and to a lesser degree in epileptogenic animals by preliminary observations). Taurine may be operating as osmoregulator counteracting the neuronal swelling and vascular alterations or as neuromodulator (of cortical synaptogenesis). Taurine concentration in the earlier phase of the disease progression (1 month postinjury, data not shown) did not correlate with the late seizure susceptibility highlighting the importance of late chronic time window for MRS scan. High macromolecular content associating with epileptogenesis may be linked with inflammatory response, plasticity related changes in extracellular matrix or degradative processes – interestingly, glia marker myoinositol, membrane turnover indicators glycerophosphocholine or total choline (or none of the other neurochemicals) in this putative future seizure onset zone did not differentiate the animals prone to epilepsy. These novel findings 1) call for clarifying the role of chronically elevated Taurine in this animal model, 2) show promise as a biomarker for the posttraumatic epileptogenesis, and 3) provide new cues for the antiepileptogenic trials.Acknowledgements
We thank J. Hartikainen, M. Lukkari and M. Pulkkinen for their expertise and technical assistance.References
1. Pitkänen A, Bolkvadze T, Immonen R. Anti-epileptogenesis in rodent post-traumatic epilepsy models. Neurosci Lett. 2011 27;497(3):163-71.
2. Bragin A, Li L, Almajano J, Alvarado-Rojas C, et al. Pathologic electrographic changes after experimental traumatic brain injury. Epilepsia. 2016 57(5):735-45.
3. Reid AY, Bragin A, Giza CC, et al. The progression of electrophysiologic abnormalities during epileptogenesis after experimental traumatic brain injury. Epilepsia. 2016 57(10):1558-1567.
4. Hayward NM, Tuunanen PI, Immonen R, et al. Magnetic resonance imaging of regional hemodynamic and cerebrovascular recovery after lateral fluid-percussion brain injury in rats. J Cereb Blood Flow Metab. 2011 31(1):166-77.
5. Kyyriäinen J, Ekolle Ndode-Ekane X, Pitkänen A. Dynamics of PDGFRβ expression in different cell types after brain injury. Glia. 2016 25. doi: 10.1002/glia.23094.
6. Liu YR, Cardamone L, Hogan RE, Gregoire MC, et al. Progressive metabolic and structural cerebral perturbations after traumatic brain injury: an in vivo imaging study in the rat. J Nucl Med. 2010 51(11):1788-95.
Figures