2937
Chronic liver disease in developing brain: an in vivo longitudinal and multiparametric study using 31P MRS, 31P Magnetization transfer and 1H MRS1Laboratory for Functional and Metabolic Imaging, EPFL, Lausanne, Switzerland, 2High Field MR Centre, Department of Biomedical Imaging and Image-Guided Therapy, Medical University of Vienna, Vienna, Austria, 3Centre d’Imagerie Biomedicale (CIBM), EPFL, Lausanne, Switzerland
Synopsis
Chronic liver disease (CLD) induces irreversible brain alterations, especially in children, probably linked with oxidative stress and energy metabolism perturbations. Our aim was to use combination of NOE enhanced and 1H-decoupled 31P-MRS, 31P-saturation transfer experiment (to estimate mitochondrial creatine kinase rate) and 1H-MRS to study the effect of CLD on developing brain. Our results show significantly reduced kATP→PCr and fluctuating NAD+/NADH ratio indicating perturbation in mitochondrial function, possibly induced by oxidative stress. In addition, altered phospholipid metabolism was observed.
Motivation
Biliary atresia is a serious condition that can occurs in children due to damaged bile duct, leading to chronic liver disease (CLD) and increased toxins in the blood (e.i. ammonia, bilirubin, bile acids, inflammatory factors) which generate brain alterations accompanied by neurocognitive deficits referred to as chronic hepatic encephalopathy (CHE). These changes are often irreversible, especially in children, and their molecular mechanisms are still not completely understood. Previous studies suggested that oxidative stress and energy metabolism perturbations can play a crucial role in pathogenesis of CHE1-4. Even though adult brain does not seem to have serious energy metabolism alterations caused by CLD5, developing brain showed stronger metabolic changes under CLD measured by 1H-MRS (among others, a stronger decrease in antioxidants compared to adults).6,7
Our aim was to perform a multiparametric study combining static and kinetic 31P-MRS and 1H-MRS for assessing the effect of CLD on developing brain in vivo and longitudinally.
Methods
Four Wistar male rats were bile duct ligated (BDL) 21 days post-natal. Another four rats underwent sham surgery at matched age to take into account the changes due to ongoing brain development. Basal temperature of each animal was carefully maintained at 37.7°C and respirations at 65-75resp./min (isoflurane anesthesia ~1.5%).
MRS studies were performed at week 2, 4 and 6 after BDL (with blood sampling) on a 9.4T-system (Varian/Magnex Scientific) using a home built dual transceive surface coil with quadrature 1H-loops and a single 31P-loop. First and second order shims were adjusted using FASTMAP8 in VOI=5x9x9mm3.
31P-MR spectra were acquired using a non-selective AHP pulse, localized by OVS (x,z) and 1D-ISIS (y). First, mitochondrial creatine kinase (MtCK) rate constant (kATP→PCr) was estimated using a 31P-saturation transfer experiment. γ-ATP was saturated with a BISTRO pulse train9 with saturation times from 0.324 to 5.184s. A control scan with irradiation offset in the mirror position relative to PCr peak was also acquired (relaxation delay=8s,80avg.). Subsequently, a non-saturated 31P-MRS scan was performed (TR=8s,384avg.) using WALTZ-16 for NOE and 1H-decoupling.
1H-MR spectra were acquired using the SPECIAL sequence10 (TE=2.8ms,TR=4s,80avg.) in the same VOI.
1H-MR spectra were quantified by LCModel using water signal as a concentration reference, 31P-MR spectra by AMARES (jMRUI)11 and normalized for each rat using its PCr concentration from 1H-MRS in the same VOI. The unequal NOE effect on different 31P metabolite peaks was corrected. High quality of 31P-spectra allowed visual distinction of intra- and extra-cellular Pi, NAD+ and NADH peaks (Fig.1). NAD+ chemical shifts measured specifically at 9.4T were used for quantification.12
Results and discussion
The presence of CLD in BDL rats was proved by a progressive increase of plasma bilirubin, highly correlated with typical increase of brain Gln (+320%) due to ammonia detoxification and a decrease of brain Ins (-37%) as an osmotic answer, measured by 1H-MRS.
BDLs showed only a slight reduction of PCr and ATP, however, kATP→PCr was significantly decreased at week 2 due to decreased enzyme activity or decreased quantity of enzyme present in cell mitochondria, signaling affected energy resources for cell (Fig.2). Later increase of kATP→PCr could be related to compensatory mechanisms, as seen with the Ins decrease. No change was observed in Piin or Piex.
tNAD pool did not change, however, a fluctuating ratio of NAD+/NADH indicates unstable cellular redox state, mainly due to variations in NADH(Fig.3). This can be linked with an increased oxidative stress and later adaptive mechanisms, Asc and GSH decrease and Lac increase measured using 1H-MRS. Though, we did not observe any significant change in intra- or extra-cellular pH.
In addition, altered phospholipid metabolism was evident already from the spectra(Fig.1). In normal postnatal brain development, the ratio of PME (PCho+PE, membrane precursors) to PDE (GPC+GPE, degradation products) should decrease (by increasing PDE).13 This was observed in our sham controls. In BDLs, PDE were markedly decreasing, mainly due to GPE, leading to increased PME/PDE ratio(Fig.4).
Conclusion
31P-MRS revealed an altered phospholipid metabolism and fluctuations in mitochondrial function, associated with CLD-induced HE in developing brain. These findings were possible due to high quality spectra offering possibility to separate NAD+ and NADH resonances and high precision in kATP→PCr estimation.
Oxidative stress may be responsible for varying cellular redox state as well as alteration in kATP→PCr. MtCK is susceptible to oxidative stress, resulting either in its impaired functions or compensatory up-regulation of its gene expression.14 Additionally, MtCK itself regulates generation of ROS.15
Phospholipid metabolism could be also altered due to increased oxidative stress by membrane lipid peroxidation.2 These changes in phospholipids seem to be characteristic for developing brain during CLD since they were not observed in adults with CLD.5
Acknowledgements
Supported by CIBM of the UNIL, UNIGE, HUG, CHUV, EPFL, the Leenaards and Jeantet Foundations. EU: FP7-PEOPLE-2012-ITN project 316679 TRANSACT.References
1. Bosoi C, et al. Systemic oxidative stress is implicated in the pathogenesis of brain edema in rats with chronic liver failure. Free Radical Biology and Medicine 2012;52(7):1228-1235.
2. Dilektasli E, at al. The effects of obstructive jaundice on the brain: An experimental study. Asian Journal of Surgery 2015;39(3):155-163.
3. Norenberg M, et al. Oxidative stress in the pathogenesis of hepatic encephalopathy. Metabolic brain disease 2004;19(3-4):313-329.
4. Norenberg M. Oxidative and nitrosative stress in ammonia neurotoxicity. Hepatology 2003;37(2):245-248.
5. Rackayova V, et al. 1H and 31P magnetic resonance spectroscopy in a rat model of chronic hepatic encephalopathy: in vivo longitudinal measurements of brain energy metabolism. Metabolic Brain Disease 2015.
6. Rackayova V, et al. Chronic Hepatic Encephalopathy in the developing and adult rat brain: an in vivo non-invasive and longitudinal metabolic investigation using 1H MRS, DTI and immunohistochemistry. Proc. Intl. Soc. Mag. Reson. Med.22 (2014).
7. Rackayova V, et al. In vivo longitudinal study of brain metabolism and edema in chronic hepatic encephalopathy in developing and adult rat brain. The 16th ISHEN meeting, London 2014.
8. Gruetter R, Tkác I. Field mapping without reference scan using asymmetric echo-planar techniques. Magn Reson Med 2000;43(2):319–323.
9. Luo Y, et al. BISTRO: an outer-volume suppression method that tolerates RF field inhomogeneity. Magn Reson Med 2001;45(6):1095-1102.
10. Mlynárik V, et al. Localized short- echo-time proton MR spectroscopy with full signal-intensity acquisition. Magn Reson Med 2006;56:965–970.
11. (http://www.mrui.uab.es/mrui)
12. Lu M, et al. Intracellular redox state revealed by in vivo 31P MRS measurement of NAD+ and NADH contents in brains. Magn Reson Med 2014;71(6):1959-1972.
13. Maddox G L. The encyclopedia of aging: a comprehensive resource in gerontology and geriatrics. Springer 2013.
14. Schlattner U, et al. Mitochondrial creatine kinase in human health and disease. Biochimica et Biophysica Acta 2006;1762(2):164-180.
15. Meyer L, et al. Mitochondrial creatine kinase activity prevents reactive oxygen species generation: Antioxidant role of mitochondrial kinase-dependent ADP re-cycling activity. Journal of Biological Chemistry 2006;281(49):37361-37371.
Figures
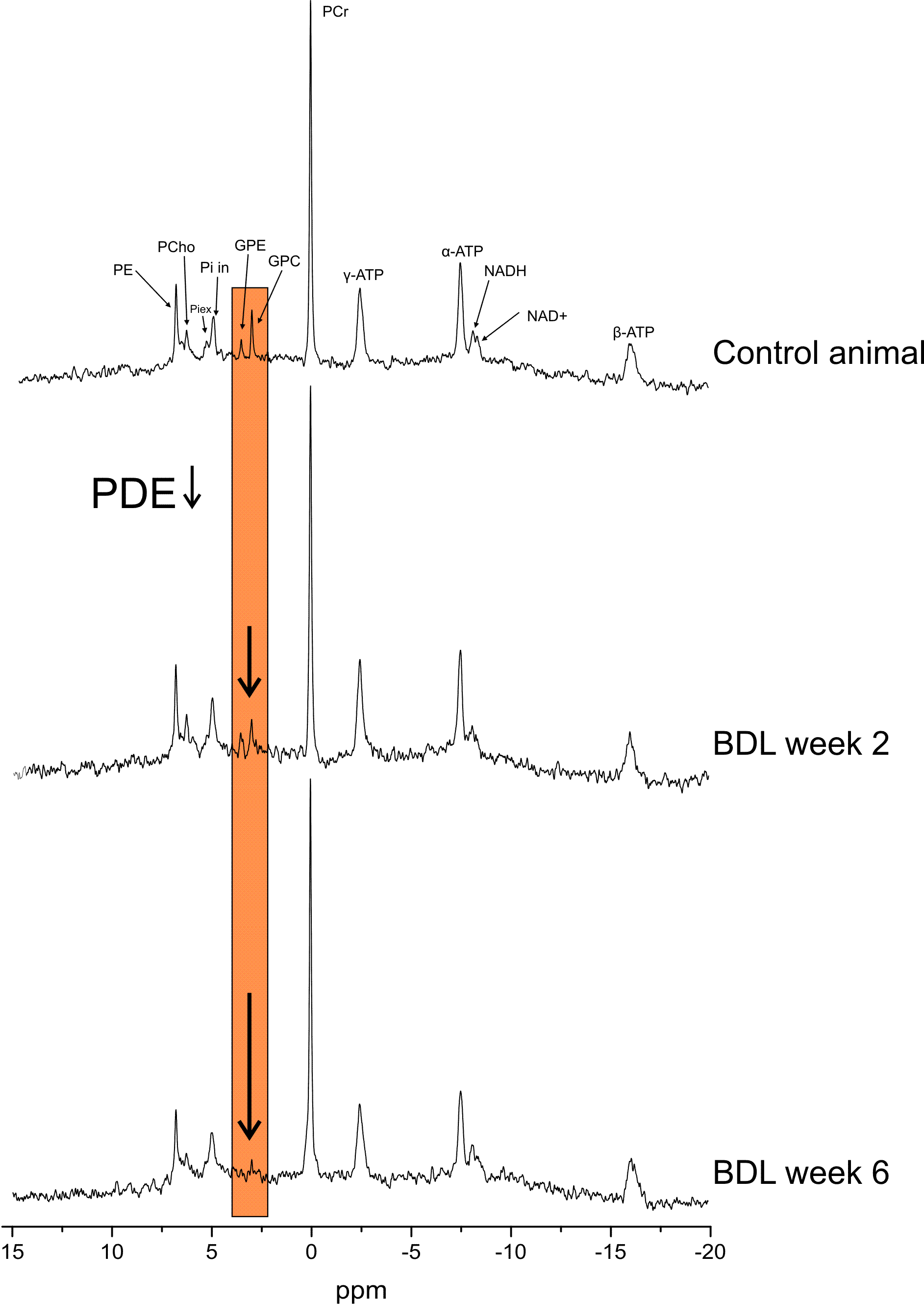
Figure 1: Non-saturated 31P-MR spectra, NOE enhanced and 1H decoupled, acquired in control sham operated animal and BDL rat 2 and 6 weeks after BDL-surgery. High SNR and spectral resolution allow visual separation of NAD+ and NADH resonances, bringing unique opportunity to asses cellular redox state in vivo and longitudinally. Strongly decreased phosphodiesters (PDE) in BDL rats are clearly observable from the spectra as the disease progresses.
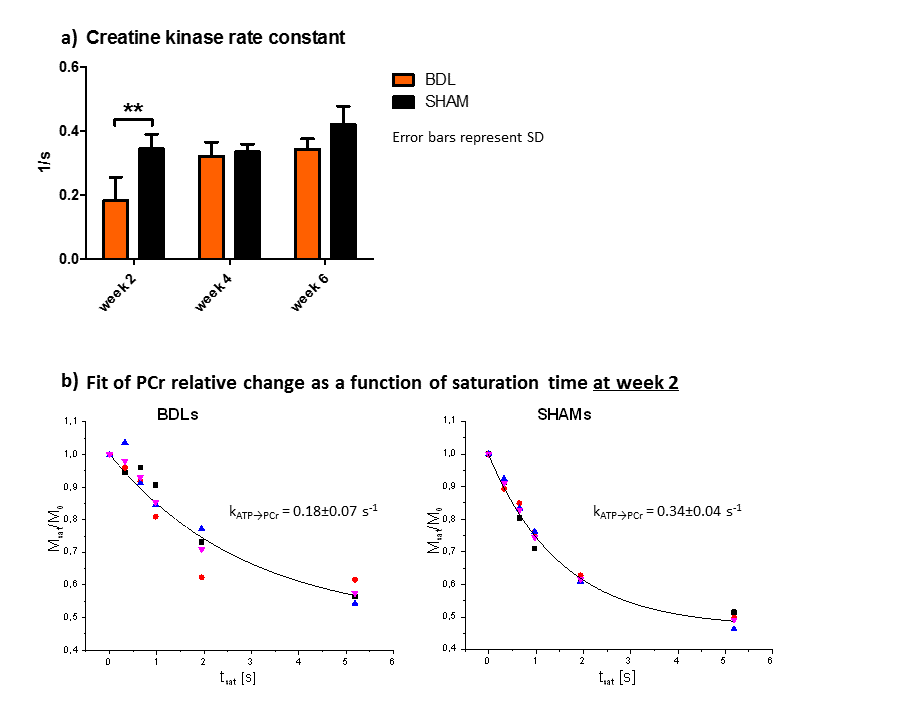
Figure 2:
a) Creatine kinase rate constant (kATP→PCr) was significantly decreased in BDL rats 2 weeks after BDL-surgery indicating perturbation in mitochondrial function.
b) Fits of relative PCr decrease due to γ-ATP saturation for BDLs and SHAMs at week 2. BDLs showed slower creatine kinase rate and also higher variability between animals.
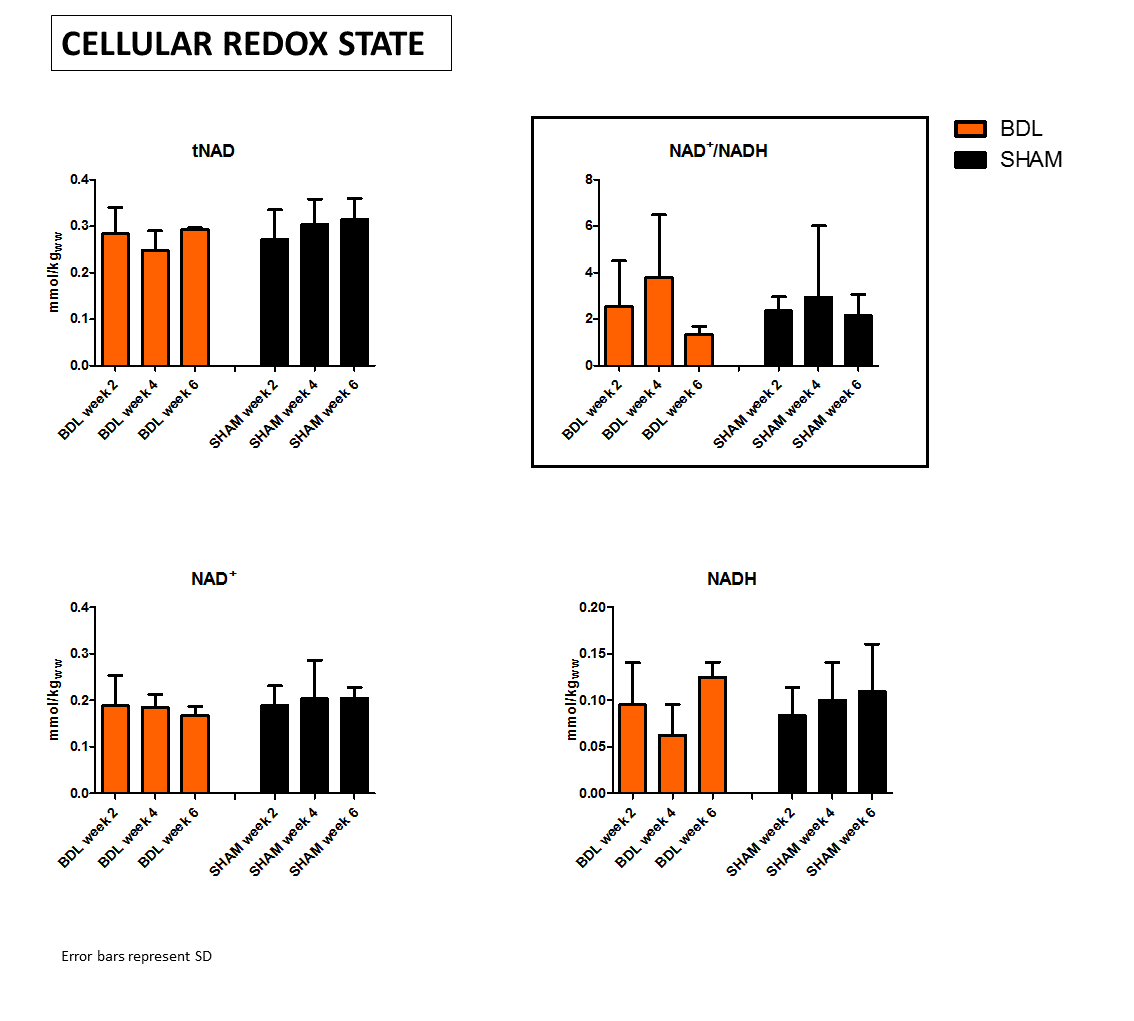
Figure 3: 31P MR spectra enabled to separate NAD+ and NADH peaks and thus estimate cellular redox state in vivo.
Total NAD pool (tNAD) represented by (NAD++NADH) did not change during CLD progression, but measurements tend to indicate variations in NAD+/NADH ratio due to variations in NADH, whitch suggests affected cellular redox state.
However, due to the small number of animals, these tendencies are not significant and further measurements are needed for confirmation.
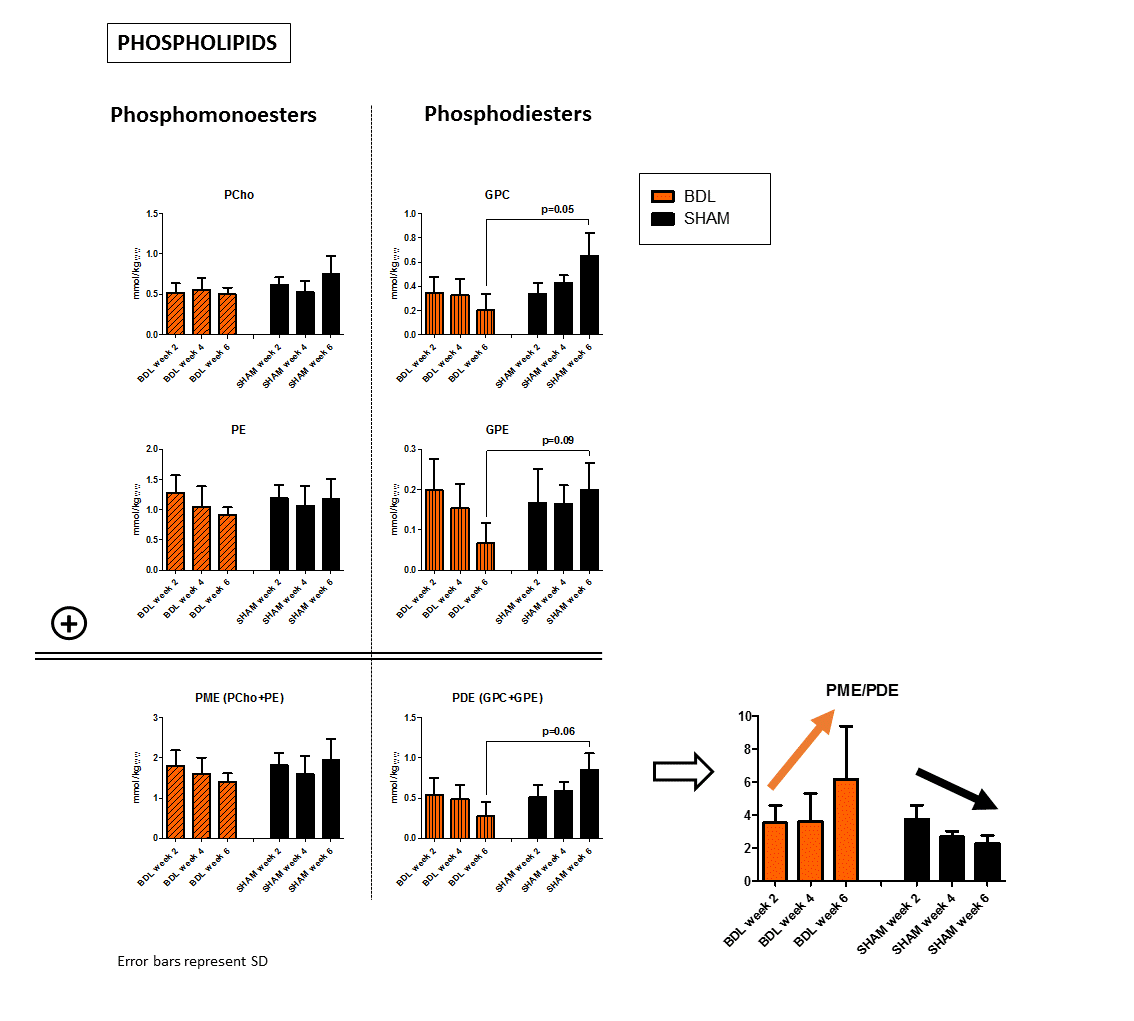
Figure 4: Four resonances are well distinguishable in brain 31P MR spectra: two phosphomonoesters (PCho, PE) representing membrane anabolism and two phosphodiesters (GPC, GPE) representing catabolic products.
Physiological increase in PDE and subsequent decrease of PME/PDE ratio due to brain development were observed in control animals. However BDLs showed reverse behavioral with decreasing PDE (especially GPE) and decreasing PME/PDE ratio. Only slight changes were observed in PME. Again, these preliminary results have to be validated with larger group of animals.