2567
MRI Characterization of Cerebellar Atrophy in a Non-Human Primate Model of Neuronal Ceroid Lipofuscinosis1Advanced Imaging Research Center, Oregon Health & Science University, Portland, OR, United States, 2Pathology, Oregon National Primate Research Center, 3Primate Genetics, Oregon National Primate Research Center, 4Oregon National Primate Research Center, 5VGTI, Oregon National Primate Research Center, 6Clinical Medicine, Oregon National Primate Research Center
Synopsis
Neuronal ceroid lipofuscinosis (NCL; also known as Batten Disease) is a fatal neurodegenerative disorder that typically presents in childhood. Currently, no treatments are known that can halt or reverse the effects of NCL. A naturally occurring form of NCL analogous to late infantile-onset NCL in humans has been identified in a population of Japanese macaques (JMs). MRI examinations revealed marked cerebellar degeneration in NCL animals >4y/o (~14y/o equivalent human age) compared with controls, which is strikingly similar to human disease. This novel JM model presents a new opportunity for characterizing disease progression, identifying biomarkers, and pre-clinical therapeutic testing.
Introduction
Neuronal ceroid lipofuscinosis (NCL; also, Batten Disease) is a fatal neurodegenerative disorder that typically presents in childhood. Symptoms include vision impairment, seizures, motor dysfunction and dementia, accompanied by cerebral atrophy and robust accumulation of storage material in the lysosomes of affected brain cells.1–3 Currently, no treatments are known that can halt or reverse the effects of NCL. However, animal models of NCL offer an opportunity to increase our understanding of underlying mechanisms and for developing new treatments for NCL patients. A naturally occurring form of NCL analogous to late infantile-onset NCL in humans has been identified in a population of Japanese macaques (Macaca fuscata, [JMs]). Clinical signs associated with the disease include ataxia, hypermetria, intention tremors, and impaired vision with onset between 4 and 6 years of age. Post-mortem findings in affected animals include marked reduction in brain size, and histology typical of NCL including cerebral and cerebellar cortical atrophy and abundant autofluorescent storage material in neurons of the CNS. The genetic basis has been identified as a single base deletion in the CLN7/MFSD8 gene. In this work, we present initial MRI findings of volumetric brain atrophy from the first known, naturally occurring non-human primate model of NCL.Methods
MRI examinations were performed on one male and five female JMs homozygous for the CLN7/MFSD8 gene defect. MRI data were available from 13 healthy control (HC) JMs. Subject demographics are detailed in Table 1. All animal care and procedures were approved by the Institutional Animal Care and Use Committee at the ONPRC. MRI data were acquired on a whole-body Siemens 3 Tesla (T) MRI instrument (Erlangen, Germany) using a quadrature radiofrequency (RF) coil with inner diameter of 15 cm. Animals were initially sedated with Telazol, intubated and maintained on 1% isoflurane in 100% O2 and were continuously monitored by pulse oximetry, respiration, and end tidal CO2 levels during the study. Quantitative R1 (≡1/T1) mapping was performed with a multiple-inversion recovery experiment detailed elsewhere.4–6 Cerebral and cerebellar volumes were calculated by a combination of linear and nonlinear co-registration (FSL)7,8 to an atlas created in-house. Masks of the cerebellum were manually corrected to ensure accuracy in all cases.Results
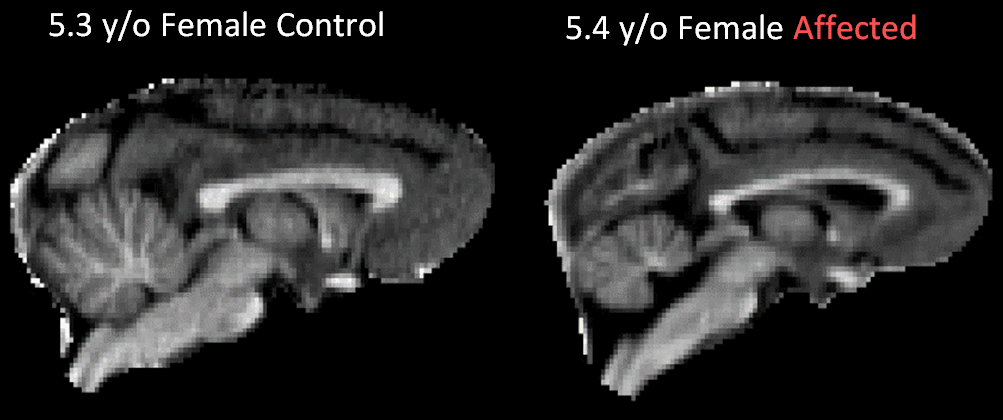
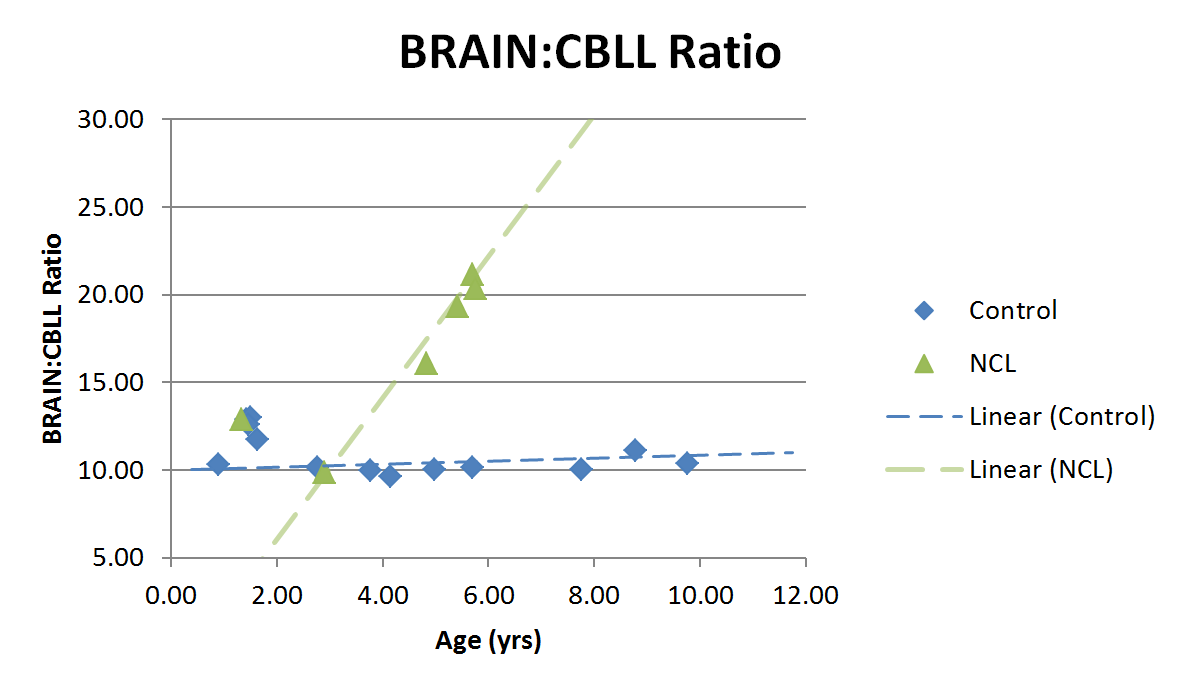
Brain volumes are inclusive of brain, cerebellum, and some brainstem; cerebellar volumes include some small portion of the peduncles, but are primarily confined to the cerebellar lobes. Midsagittal views of qR1 maps for one NCL and one age- and sex-matched control are shown in Figure 1. The calculated Brain:CBLL volume ratio (whole brain volume divided by cerebellar volume) is plotted as a function of age in Figure 2. Linear trend lines are added to guide the eye. Relative cerebellar volume in NCL-positive animals appears within a normal range until at least 2 y/o; at some point after that the disease causes cerebellar atrophy, which is notable by 5 y/o and progresses rapidly with age. Necropsy findings from four animals revealed generalized reduction in brain size, with cerebellum and occipital lobes appearing most affected. There was corresponding reduction in brain weight (mean 69.8g). This is in contrast to the average brain weight of female Japanese macaques 4 years of age and older of 101.4 g (from 168 individuals).Discussion
We found that the affected animals show marked cerebral and cerebellar cortical atrophy, with a concomitant increase in the ventricular space containing the cerebral spinal fluid. These changes are similar to that seen in human patients with a CLN7 mutation. Importantly, the timeline of disease-related morphological changes tracks well with what is seen in humans with juvenile NCL: MRI is normal in patients <10 y/o, then cerebellar atrophy becomes significant around age 141 (~4.5 y/o equivalent JM age). The variable and slightly elevated volume ratios before 2 years of age may reflect some natural variability during early development. Because we have only presented volume ratios, Figure 2 likely represents cerebellar atrophy outpacing cerebral atrophy due to disease in NCL. Future directions for MRI analysis include quantitative analysis of volumetrics and R1 changes in cortex, white matter, and deep gray matter structures in NCL versus controls to provide a more comprehensive comparison of JM NCL and human disease.Conclusions
The JM model of CLN7 NCL recapitulates human NCL disease. The JM model presents a new opportunity for characterizing disease progression, identifying biomarkers and pre-clinical therapeutic testing. We are currently screening our JM colony to identify all undiagnosed affected individuals and CLN7 carriers. Additional pathologic, genetic, and MRI analyses are ongoing. Opportunities to establish targeted breeding group to increase models for study will be of great interest and are being investigated.Acknowledgements
Funding: National Institutes of Health Grant [P51 OD011092]References
1. Williams, R. E. et al. Diagnosis of the neuronal ceroid lipofuscinoses: An update. Biochim. Biophys. Acta - Mol. Basis Dis. 1762, 865–872 (2006).
2. Jadav, R. H. et al. Clinical, electrophysiological, imaging, and ultrastructural description in 68 patients with neuronal ceroid lipofuscinoses and its subtypes. Pediatr. Neurol. 50, 85–93 (2014).
3. Haltia, M. The neuronal ceroid-lipofuscinoses: From past to present. Biochimica et Biophysica Acta - Molecular Basis of Disease 1762, 850–856 (2006).
4. Rooney, W. D. et al. MRI Estimation of Sub-Clinical Disease in Japanese Macaque Encephalomyelitis. in Proc Int Soc Magn Reson Med 17, 6973 (2009).
5. Tagge, I. et al. Blood-Brain-Barrier Permeability and Lesion Volume Changes in Acute Japanese Macaque Encephalomyelitis. Proc Int Soc Magn Reson Med 23, 238 (2015).
6. Tagge, I. et al. Lesion Distribution Probability in Japanese Macaque Encephalomyelitis: A Comparison to Human Demyelinating Diseases. in Proc Int Soc Magn Reson Med 1319 (2016).
7. Woolrich, M. W. et al. Bayesian analysis of neuroimaging data in FSL. Neuroimage 45, S173-86 (2009).
8. Jenkinson, M., Beckmann, C. F., Behrens, T. E. J., Woolrich, M. W. & Smith, S. M. Fsl. Neuroimage 62, 782–90 (2012).
Figures