2460
Reduced cortical thickness in patients with sickle cell disease and a high pain burden: baseline results from the Prevention of Morbidity in Sickle Cell Anaemia trial1Developmental Neurosciences, UCL Great Ormond Street Institute of Child Health, London, United Kingdom, 2School of Psychology, University of Southampton, Highfield, United Kingdom
Synopsis
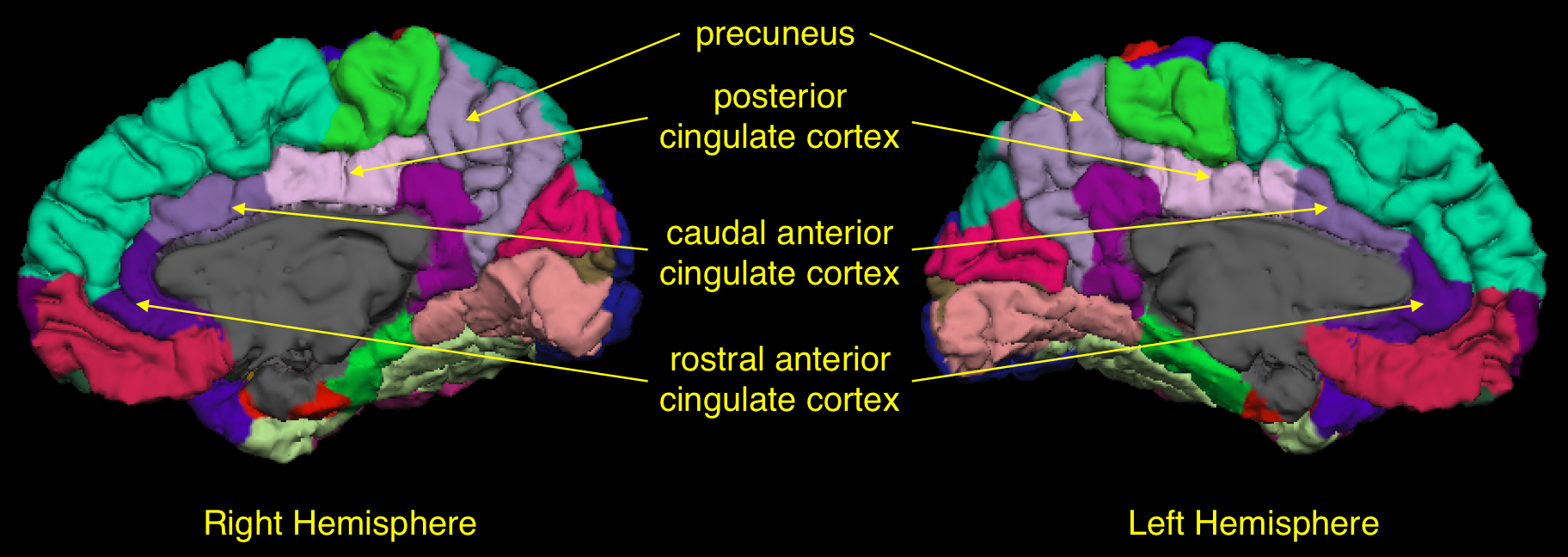
Although acute vaso-occlusive pain crises are common in sickle cell disease (SCD), some patients also experience chronic daily pain. This study investigated cortical areas involved in pain processing in low-pain and high-pain groups of patients at baseline of a trial with a pain burden outcome. High-pain patients had significantly thinner cortex in the right anterior cingulate cortex, bilateral posterior cingulate cortex, bilateral precuneus and left primary motor cortex. This is the first study showing structural brain abnormalities in patients with SCD and a high pain burden; these data may provide potential biomarkers for longitudinal trials of treatment for chronic pain.
Purpose
Sickle cell anaemia (SCA) is associated with frequent episodes of vaso-occlusive pain crises, which are the most common reason for hospitalisation in this population. While acute pain crises are common, many patients also experience chronic daily pain1, which may be due to avascular necrosis of joints, bone infarction, leg ulcers and chronic osteomyelitis or due to intractable chronic pain without obvious pathology2,3. Children with SCA as young as 6 months can experience pain4; yet, not all patients experience a significant chronic pain burden5. The impact of chronic sickle cell pain on the brain is not yet clear; a recent resting-state fMRI study showed a correlation between functional connectivity of the default-mode network and number of hospitalizations for pain6. In other chronic pain conditions, reduced grey matter volume has been noted in various regions involved in pain processing such as the thalamus7, hippocampus7–9, amygdala6,7, anterior7,10,11 and posterior6–13 cingulate cortex (ACC and PCC, respectively), primary motor cortex13,14 (M1), primary somatosensory cortex6–9,13 (S1) and insula cortex. The aim of this study is to investigate the relationship between daily pain and a priori selected grey matter regions in a sample of children and adults with SCA.Methods
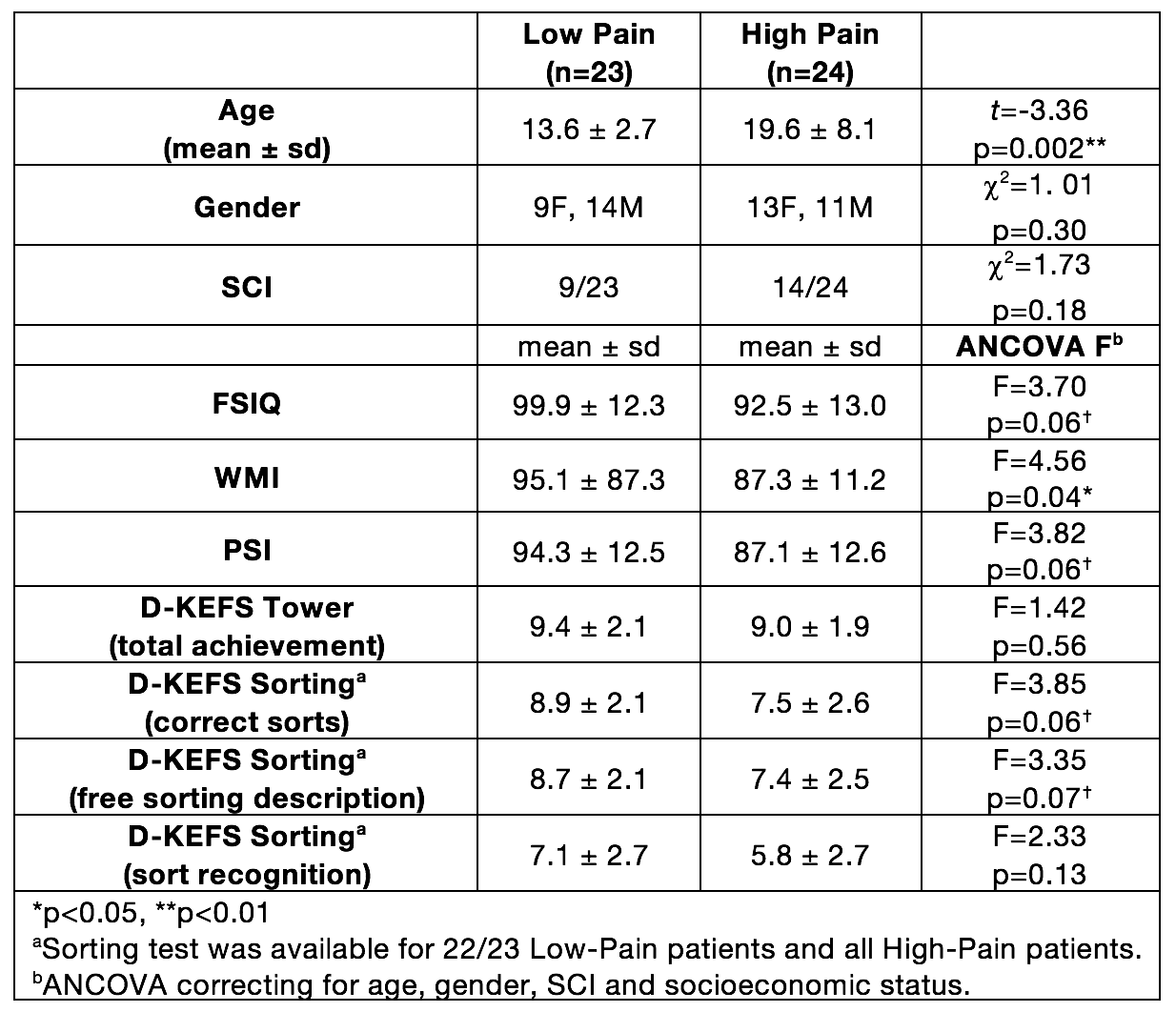
As part of the baseline assessments for the Prevention of Morbidity in Sickle Cell Anaemia (POMS) phase-II trial, 52 children and adults recorded baseline daily pain and underwent neuropsychological testing and MRI in the 14 days prior to randomisation to the trial. Patients were excluded from the trial if he/she had a hospital admission for pain within one month. Daily pain was recorded on a numerical scale 0-10 for 14 days. Patients were split into “Low-Pain” group (<50% recorded days in pain) or “High-Pain” group (>50% of recorded days in pain). Average pain for the previous 24 hours (0-10) and current pain (0-10) was recorded and averaged over all recorded days. Neuropsychological assessment was carried out using: Wechsler Abbreviated Scale of Intelligence (WASI) 2-subtest measure of full-scale IQ (FSIQ), Wechsler Abbreviated Scale of Intelligence (WAIS-IV) working memory index (WMI) and processing speed index (PSI), Delis-Kaplan Executive Function System (D-KEFS) Tower and Sorting subtests. Patients underwent MRI on a 3T Siemens Prisma (Erlangen, Germany) that included an axial T2-weighted sequence (TR=8420ms, TE=68ms, voxel size=0.51x0.51x5.6mm) and coronal FLAIR sequence (TR=5000ms, TE=395ms, voxel size=0.65x1x0.65mm) to diagnose presence or absence of silent cerebral infarction and a 3D T1-weighted MPRAGE (TR=2300ms, TE=2.74ms, voxel size=1mm3). Cortical parcellation and subcortical segmentation was carried out using Freesurfer v5.3 (http://surfer.nmr.mgh.harvard.edu/). A model was fitted to the imaging data variables to investigate differences between low-pain and high-pain groups, controlling for age, gender and presence of SCI15. Differences in cognitive measures between low-pain and high-pain groups used age, gender, SCI16 and socioeconomic status17 (Index of Multiple Deprivation decile by UK postcode) as covariates.Results
Five children were excluded from analysis due to excessive motion, poor quality data or braces artefact. The final sample was 47 children and adults with SCA (Figure 1). The high-pain group was significantly older than the low-pain group and there was a significant correlation with percentage of days in pain and age (r=0.53, p=0.0001). In the low-pain group, 11 patients reported no pain during the recorded days. Patients in the high-pain group had significantly lower WMI and a trend for lower FSIQ, PSI, Sorting – correct sorts and Sorting – free sorting description (Figure 1). High-pain patients had significantly reduced cortical thickness in the mean cortex of the left and right hemispheres, and also significantly thinner cortex in the bilateral precuenus, bilateral PCC, left M1 and right caudal ACC (Figure 2). There was a trend for positive correlation between average pain and right-hemisphere rostral ACC thickness. There were no significant differences in volumes of the bilateral thalamus, bilateral hippocampus and bilateral amygdala between low- and high-pain groups.Conclusion
This study is the first to describe structural brain abnormalities in patients with high burden of sickle cell pain. Several cortical areas involved in pain processing that underlie the default-mode network6 were found to be significantly thinner in high-pain patients. These patients are part of a 6-month randomised controlled trial of overnight oxygen support with cognitive and pain endpoints as well as repeat MRI; this study may provide structural biomarkers that may parallel amelioration of chronic daily pain with interventions including reduction of hypoxic exposure18.Acknowledgements
No acknowledgement found.References
1. Ballas SK, Gupta K, Adams-Graves P. Sickle cell pain: a critical reappraisal. Blood 2012;120(18):3647–3656.
2. Ballas SK. Pain Management of Sickle Cell Disease. Hematol. Oncol. Clin. North Am. 2005;19(5):785–802.
3. Benjamin LJ, Payne R. Pain in sickle cell disease: a multidimensional construct. In: Pace P, editor. Renaissance of Sickle Cell Disease Research in the Genome Era. London, United Kingdom: Imperial Press; 2007 p. 99–116.
4. Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease. Rates and risk factors. N. Engl. J. Med. 1991;325(1):11–6.
5. Dampier C, Ely B, Brodecki D, O[apos ]Neal P. Characteristics of pain managed at home in children and adolescents with sickle cell disease by using diary self-reports. J. Pain 2002;3(6):461–470.
6. Darbari DS, Hampson JP, Ichesco E, et al. Frequency of Hospitalizations for Pain and Association With Altered Brain Network Connectivity in Sickle Cell Disease. J. Pain 2015;16(11):1077–1086.
7. De Kruijf M, Bos D, Huygen FJPM, et al. Structural brain alterations in community dwelling individuals with chronic joint pain. Am. J. Neuroradiol. 2016;37(3):430–438.
8. Hubbard CS, Khan SA, Keaser ML, et al. Altered Brain Structure and Function Correlate with Disease Severity and Pain Catastrophizing in Migraine Patients. eNeuro 2014;1(1):2–17.
9. Liu P, Yang J, Wang G, et al. Altered regional cortical thickness and subcortical volume in women with primary dysmenorrhoea. Eur. J. Pain (United Kingdom) 2016;20(4):512–520.
10. Mordasini L, Weisstanner C, Rummel C, et al. Chronic Pelvic Pain Syndrome in Men is Associated with Reduction of Relative Gray Matter Volume in the Anterior Cingulate Cortex Compared to Healthy Controls. J. Urol. 2012;188(6):2233–2237.
11. Younger JW, Shen YF, Goddard G, Mackey SC. Chronic myofascial temporomandibular pain is associated with neural abnormalities in the trigeminal and limbic systems. Pain 2010;149(2):222–228.
12. Ceko M, Bushnell MC, Fitzcharles MA, Schweinhardt P. Fibromyalgia interacts with age to change the brain. NeuroImage Clin. 2013;3:249–260.
13. Hubbard CS, Becerra L, Heinz N, et al. Abdominal pain, the adolescent and altered brain structure and function. PLoS One 2016;11(5):1–30.
14. Moayedi M, Weissman-Fogel I, Crawley AP, et al. Contribution of chronic pain and neuroticism to abnormal forebrain gray matter in patients with temporomandibular disorder. Neuroimage 2011;55(1):277–286.
15. Kawadler JM, Clayden JD, Kirkham FJ, et al. Subcortical and cerebellar volumetric deficits in paediatric sickle cell anaemia. Br. J. Haematol. 2013;163(3):373–6.
16. Kawadler JM, Clayden JD, Clark CA, Kirkham FJ. Intelligence Quotient in Paediatric Sickle Cell Disease: a Systematic Review and Meta-Analysis. Dev. Med. Child Neurol. 2016;
17. King AA, Strouse JJ, Rodeghier MJ, et al. Parent education and biologic factors influence on cognition in sickle cell anemia. Am. J. Hematol. 2014;89(2):162–7.
18. Hargrave DR, Wade A, Evans JPM, et al. Nocturnal oxygen saturation and painful sickle cell crises in children. Blood 2003;101(3):846–8.
Figures