2237
Altered default mode network organization and functional connectivity in mice lacking autism-associated gene Shank31Functional Neuroimaging Laboratory, Center for Neuroscience and Cognitive Systems, Istituto Italiano di Tecnologia, Rovereto, Italy, 2CIMeC, Center for Mind/Brain Sciences, University of Trento, Rovereto, Italy
Synopsis
Mutations in autism-associated gene Shank3 have been associated to alterations in striatal function and core autistic behaviors. However, the neocortical substrates affected by Shank3 mutations remain undetermined. By using structural and functional MRI in Shank3B mutant mice, we identified key alterations in prefrontal and associative regions of the mouse default mode network (DMN). Specifically, we show that prefrontal and antero-posterior areas of the DMN present decreased gray matter volume, an effect associated with reduced local and long-range prefrontal functional connectivity. Our findings suggest that Shank3 mutations may predispose to autism via a selective trophic and functional downregulation of prefrontal areas.
BACKGROUND
Functional connectivity aberrancies, as measured with resting-state fMRI (rsfMRI), have been consistently observed in the brain of autism spectrum disorders (ASD) patients1. ASD has a strong genetic etiology and mouse models recapitulating human disease-causing mutations can be used to establish causal links between ASD-related genetic etiologies and abnormal macroscale connectivity, thus shedding light on the connectional underpinnings of this phenomenon2. Shank3 encodes for a key postsynaptic density protein at glutamatergic synapses whose mutation has been associated with core language and/or social communication impairments in humans ASD-patients3. Mouse studies have highlighted altered molecular composition of striatal synapses and morphological defects of medium spiny neurons as well as ASD-like repetitive grooming and deficits in mice lacking the Shank3B isoform4. However, despite compelling evidence of cognitive impairments in human carriers of Shank3 mutations and Shank3B mutant mice5, the neocortical substrates affected by Shank3 mutations remain undetermined. To address this question, we used high resolution structural MRI6 and in vivo resting-state fMRI (rsfMRI)7,8, to probe gray matter volume and neocortical functional connectivity in Shank3B-/- mice4. In doing so, we identified previously unreported focal structural and functional alterations in the default mode network of Shank3B mutants, which serve as a plausible neuro-functional correlate for the cognitive impairments observed in this mouse line5.METHODS
All experiments were carried out in accordance with Italian
regulations governing animal welfare and protection. Mice (Shank3B-/-, n=11 and control littermates,
n=10) were imaged at 7 tesla under
shallow halothane anesthesia (0.75%) and artificial ventilation8,9 using a single-shot EPI sequence (TR/TE 1200/15 ms, flip angle 30°, FOV 2×2 cm2, matrix 100×100, 24 coronal slices, slice thickness 0.50 mm, 500
volumes). Data were preprocessed as previously described7. We calculated global and local brain connectivity maps for all subjects
and mapped voxelwise inter-group differences in both measures and additionally
mapped anteroposterior DMN connectivity between a series of default mode
network seeds. We also
employed post-mortem voxel based morphometry (VBM) and automated anatomical
labelling (AAL)10 on high resolution images obtained with a FLASH
sequence with an isotropic voxel size of 70 µm. The same brains underwent DTI
imaging using 80 directions, a 120x120x240 voxel size, and b=3000, d=6 and D=13
ms as recently described11,12.RESULTS AND DISCUSSION
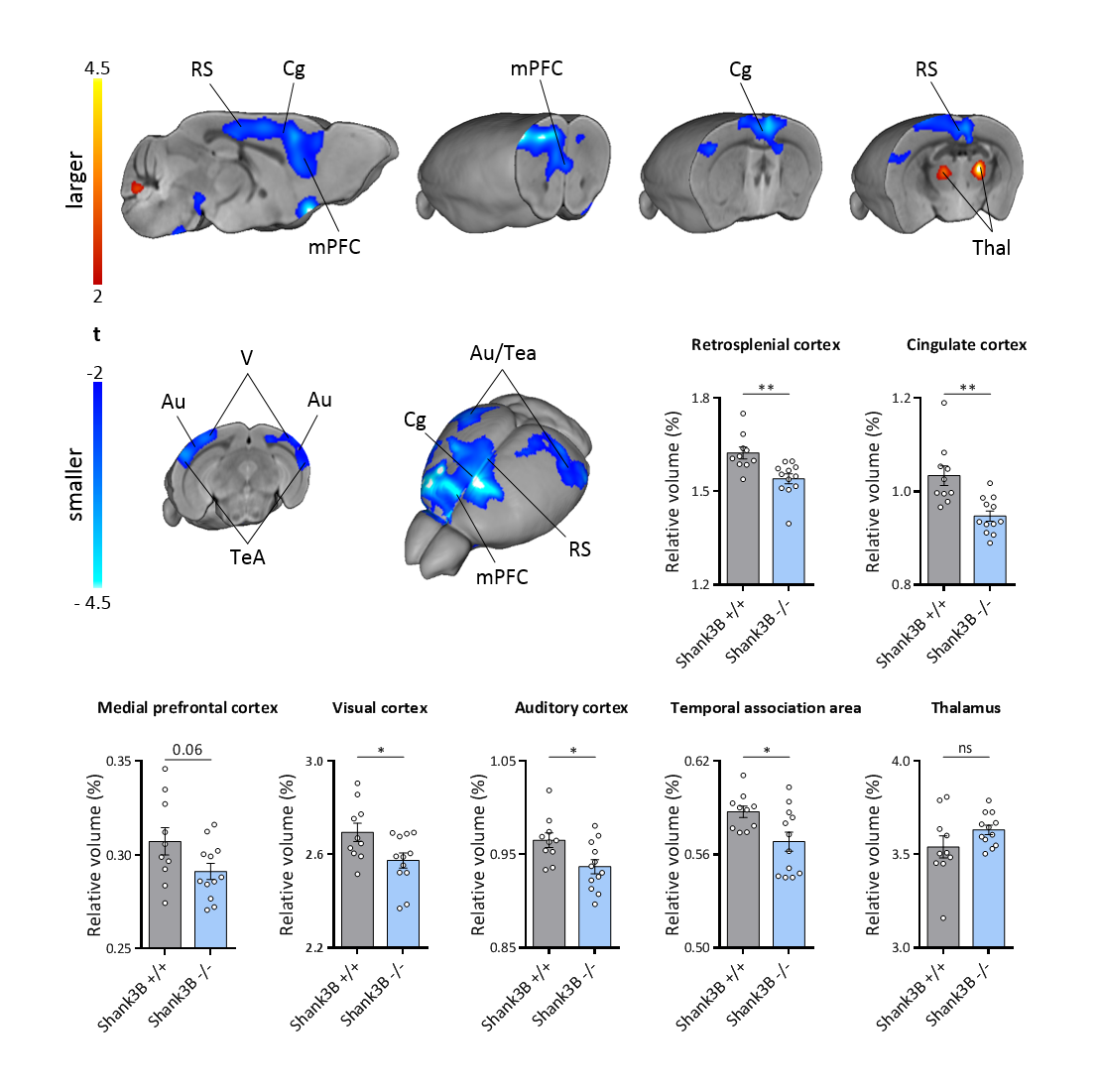
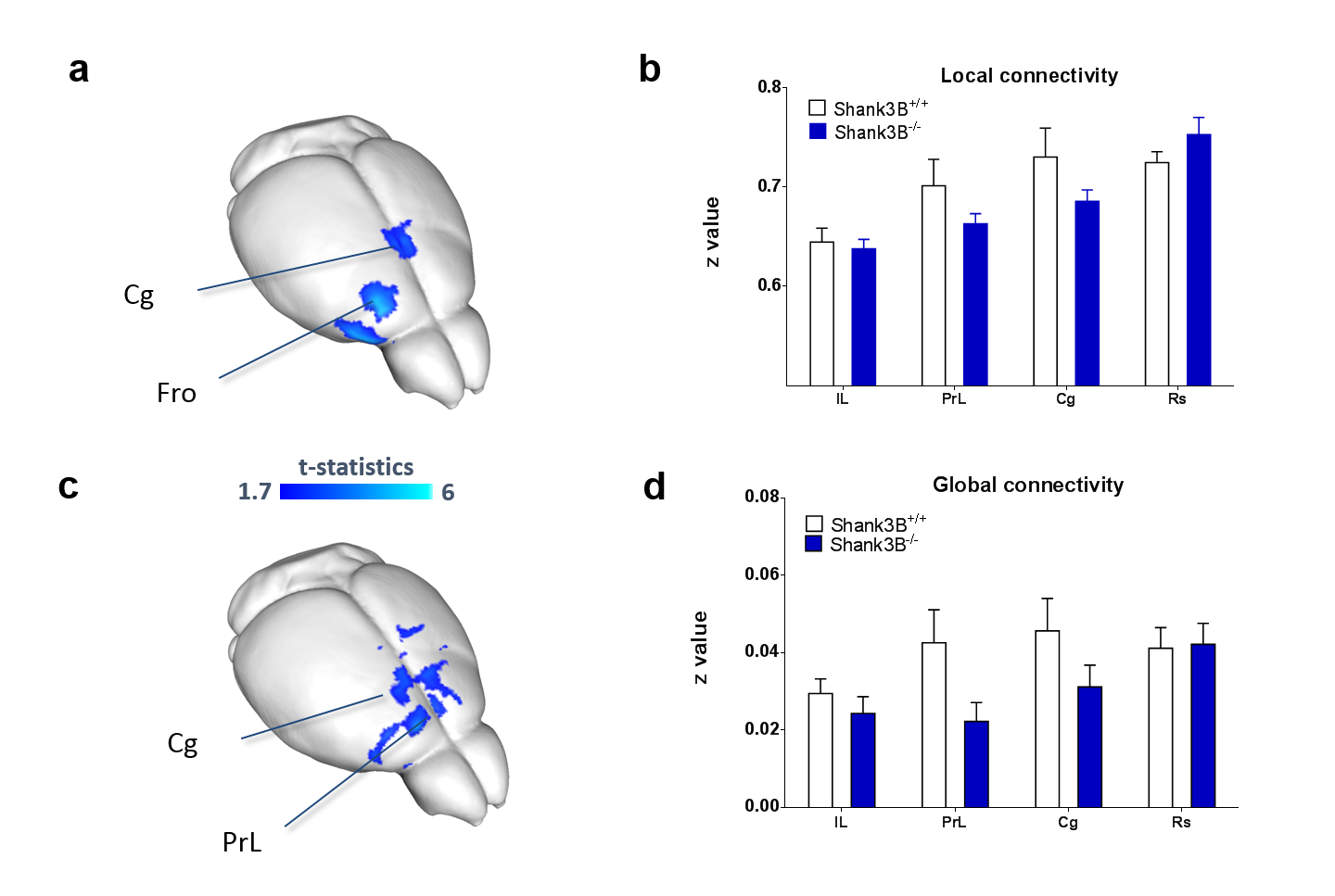
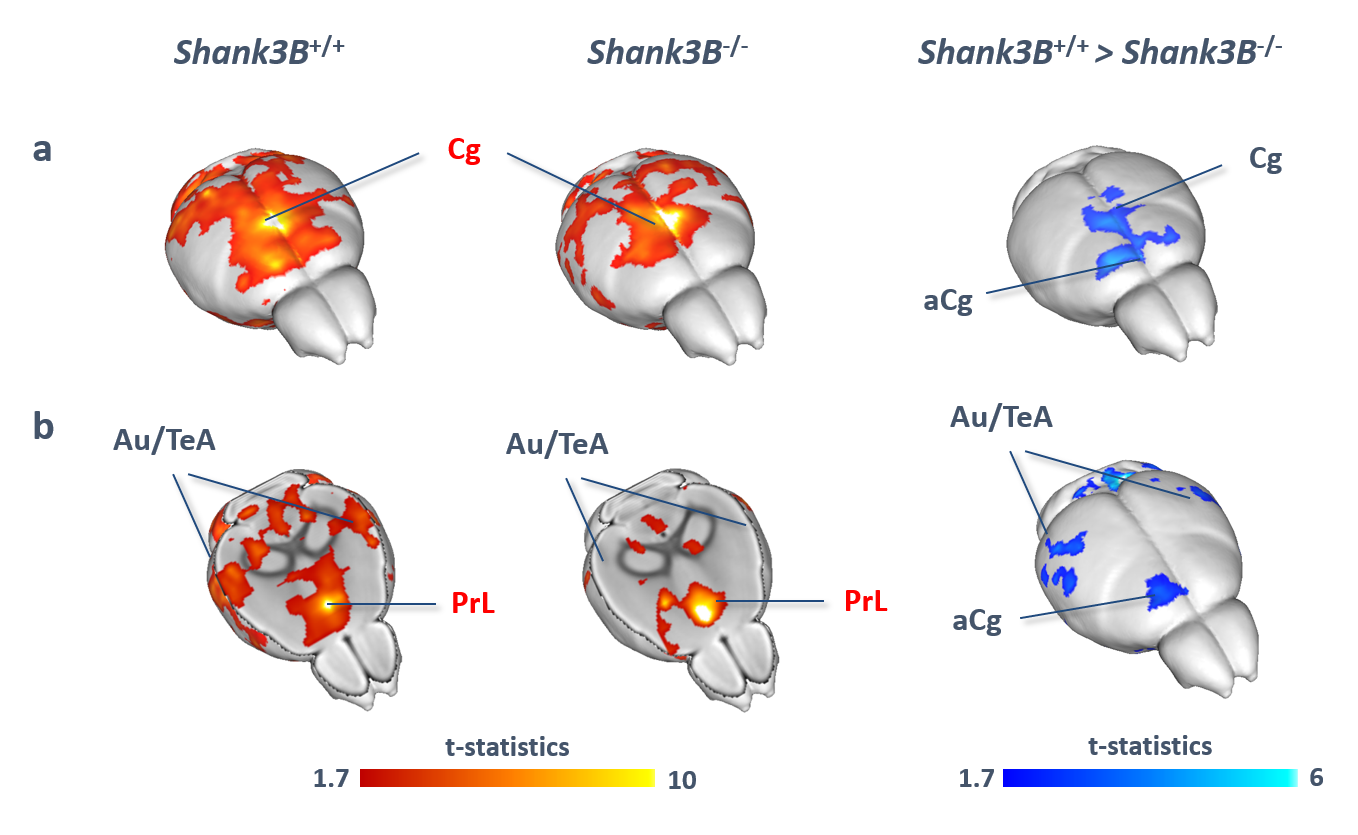
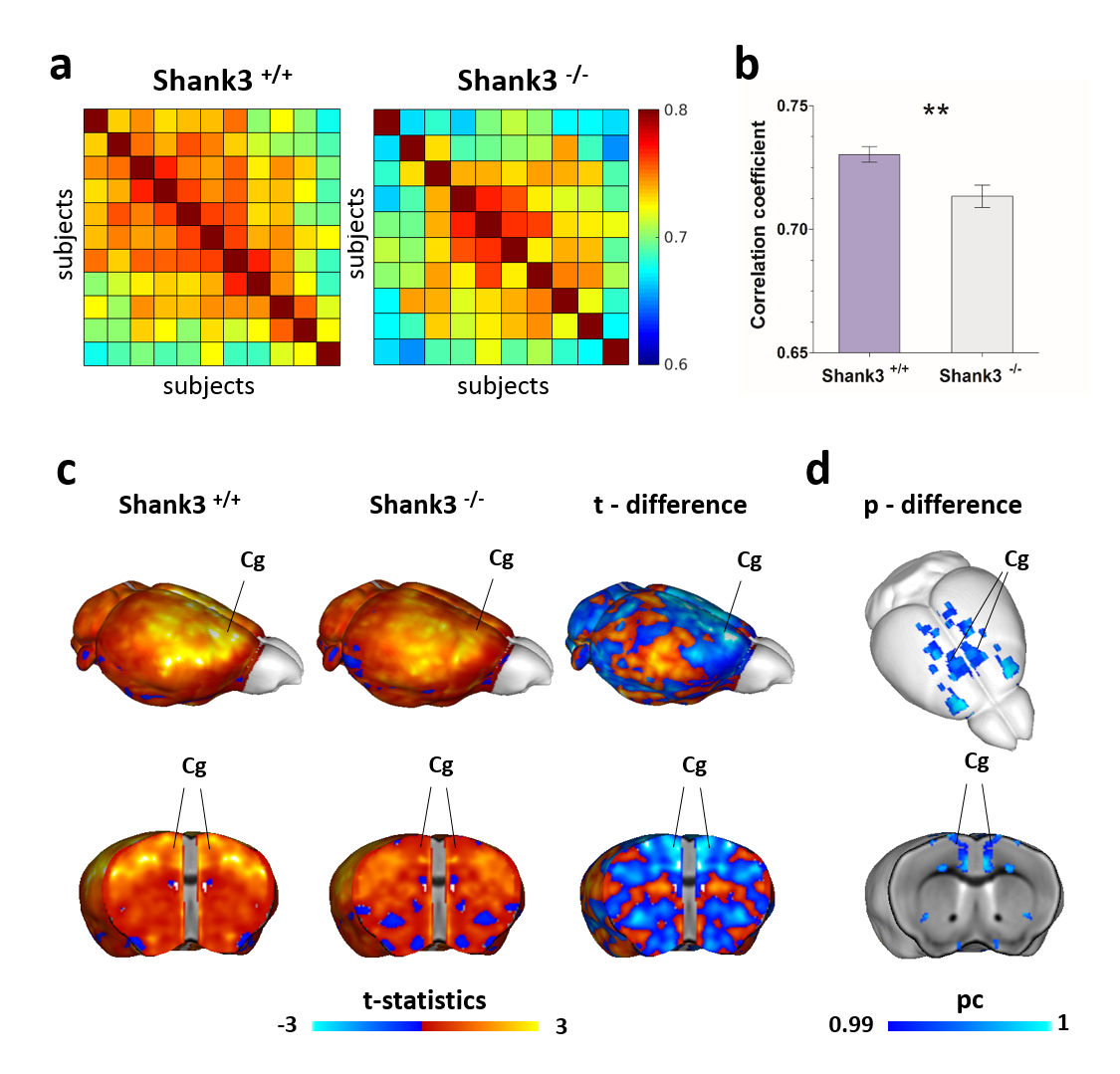
VBM and AAL revealed foci of gray matter increase in thalamic areas, along with broad and prominent reductions in gray matter volume in Shank3B-/- mice encompassing a network of cortical regions exhibiting remarkable neuroanatomical resemblance with key the prefrontal, midline and temporal nodes of the mouse default mode network (Fig.1). This finding led us to postulate the presence of reduced functional coupling between these regions. Spatially unbiased local and global connectivity mapping revealed a significant rsfMRI connectivity reduction in anterior cingulate regions (Fig.2). To understand whether this effect could partly reflect antero-posterior DMN dysconnectivity, we performed seed-based correlation mapping. This analyses revealed hypo-connectivity in middle and anterior cingulate regions, as well as a reduced involvement of temporal cortical areas (belonging to the mouse DMN) (Fig.3). These findings suggest the presence of weak but focal functional hypo-connectivity in prefrontal and associative cortical regions of the DMN, consistent with reduced gray matter trophism in the same areas (Fig.1). The observation of inter-subject inhomogeneous expression of GABAergic neuronal markers in mouse models of autism13 led us to postulate the presence of highly variable, “idiosyncratic” homotopic connectivity in these mice. Interestingly, Shank3B mutants exhibited decreased inter-subject similarity in Shank3B group compared to controls (Fig.4), suggesting the presence of idiosyncratic connectivity patterns in mutant mice and the presence of a ‘regression to the mean’ effect3 reminiscent of analogous findings recently reported in human ASD patients. No apparent genotype-dependent alterations in white macrostructure or fiber-organization were observed as seen with DTI-based fractional anisotropy mapping, or tractographic analyses, thus supporting the notion of functional (rather than neuro-connectional) origin of the alterations mapped.CONCLUSION
We show that Shank3B deletion leads to cortical volumetric loss and reduced local and long-range functional connectivity within prefrontal and posterior hub regions of the mouse default mode network. These findings reveal a plausible neuro-functional correlate for the cognitive impairments observed in this mouse line, and suggest that Shank3 mutations may predispose to autism via a selective trophic and functional downregulation of prefrontal areas.Acknowledgements
The study was funded by a grant from the Simons Foundation (SFARI 314688 and 400101, A.G.)References
1. Anagnostou, E. and M.J. Taylor, Review of neuroimaging in autism spectrum disorders: what have we learned and where we go from here. Molecular autism, 2011. 2(1): p. 1.
2. Liska, A. and A. Gozzi, Can mouse imaging studies bring order to autism connectivity chaos? Frontiers in Neuroscience, 2016. 10(484).
3. Durand, C.M., et al., Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nature genetics, 2007. 39(1): p. 25-27.
4. Peça, J., et al., Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature, 2011. 472(7344): p. 437-442.
5. Copping, N.A., et al., Touchscreen learning deficits and normal social approach behavior in the Shank3B model of Phelan–McDermid Syndrome and autism. Neuroscience, 2016.
6. Pagani, M., et al., Semi-automated registration-based volumetric labelling, voxel based morphometry and cortical thickness mapping of the mouse brain. Submitted, 2015.
7. Liska, A., et al., Functional connectivity hubs of the mouse brain. NeuroImage, 2015. 115(0): p. 281-291.
8. Sforazzini, F., et al., Altered functional connectivity networks in acallosal and socially impaired BTBR mice. Brain Struct Funct, 2014: p. 1-14.
9. Sforazzini, F., et al., Distributed BOLD and CBV-weighted resting-state networks in the mouse brain. NeuroImage, 2014. 87: p. 403-415.
10. Pagani, M., et al., Semi-automated registration-based anatomical labelling, voxel based morphometry and cortical thickness mapping of the mouse brain. Journal of neuroscience methods, 2016. 267: p. 62-73.
11. Dodero, L., et al., Neuroimaging evidence of major morpho-anatomical and functional abnormalities in the BTBR T+ TF/J mouse model of autism. PloS one, 2013. 8(10): p. e76655.
12. Liska, A., et al., Homozygous loss of autism-risk gene CNTNAP2 results in reduced local and long-range prefrontal functional connectivity. bioRxiv, 2016.
13. Gogolla, N., et al., Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J Neurodev Disord, 2009. 1(2): p. 172-181.
Figures