0613
Striatal Glutathione Deficit in Parkinson’s Disease Measured In Vivo with J-edited 1H MRS Directly Implicates Oxidative Stress in Disorder Pathophysiology1Radiology, Weill Cornell Medicine, New York, NY, United States, 2Neurology and Neuroscience, Weill Cornell Medicine, New York, NY, United States, 3Healthcare Policy and Research, Weill Cornell Medicine, New York, NY, United States
Synopsis
Postmortem studies of Parkinson’s disease (PD) brain have consistently reported deficits of nigrostriatal glutathione (GSH) – the most abundant antioxidant in living tissue – of up to 40% compared to normal brain, strongly implicating oxidative stress in the pathophysiology of PD. However, direct evidence corroborating a striatal GSH deficit in PD brain in vivo is currently lacking. Using J-edited 1H MRS, this study measured striatal GSH in vivo in patients with PD and in matched control subjects, and found not only a 15% deficit of striatal GSH in PD that corroborated postmortem data, but also evidence of nigrostriatal neurodegeneration in the disorder.
INTRODUCTION
Multiple lines of evidence, including findings of membrane lipid and protein peroxidation/nitrosylation and oxidative DNA damage1, have implicated oxidative stress in the chain of pathogenic events that lead to the progressive degeneration of nigrostriatal dopaminergic neurons in Parkinson’s disease (PD)2 – the hallmark of the disorder. Importantly, replicated postmortem PD brain studies3-6 have consistently found loss of nigral glutathione (GSH) – the primary living tissue antioxidant – of up to 40%, further implicating oxidative stress in the disorder.7 However, in vivo human brain evidence corroborating this postmortem GSH deficit is currently lacking. The objective of this pilot study was to assess whether in vivo levels of striatal GSH are lower in patients with PD than in matched healthy control subjects, as suggested by postmortem data3-6, by using J-edited 1H MRS to measure GSH levels directly and noninvasively in living PD and normal brain.
METHODS
Subjects: For this clinical study, 22 patients (mean age: 61±5yr; 6 females) diagnosed with idiopathic PD per the United Kingdom Parkinson’s Disease Society Brain Bank criteria (UKPDSBB) criteria, and 27 medically healthy volunteer (HV) subjects (mean age: 60±7yr; 5 females) were recruited. Patients with PD were excluded if they were taking dopamine receptor blocking agents; had major depression or a concomitant medical disease limiting life expectancy to less than 12 months from study inclusion; had a diagnosis of primary mitochondrial disorder, epilepsy, stroke, multiple sclerosis or neurodegenerative diseases; and had Mini-Mental Status Exam (MMSE) score ≤ 24/30 or a disease duration ≥15yr. Study procedures included a complete medical examination and J-edited 1H MRS to measure levels of GSH and other neurometabolites in the striatum (primary region of interest) and in the occipital cortex (OCC) as a “control” region not implicated in PD.
1H MRS of GSH: In vivo spectra of GSH and other major neurometabolites were recorded in 15 min on a 3T GE MR system from a 2.0x3.0x2.0-cm3 striatal voxel (Figure 1A) and a 2.0x3.0x3.0-cm3 OCC voxel (Figure 1B), using J-edited 1H MRS (Figure 1C) and an 8-channel phased-array head coil, with TE/TR 68/1500ms and 290 interleaved excitations. Levels of all neurometabolites, including GSH, were derived by frequency-domain fitting (see Figure 1C for GSH fitting), and then expressed as peak area ratios relative to the unsuppressed intravoxel water (W) signal. General linear models were used to compare mean neurometabolite levels between PD patients and HV subjects.
RESULTS
In the striatum, the region of primary interest, GSH/W in PD patients ([3.5±0.1]x10-3) was significant lower (p=0.04) than in the HV group ([4.1±0.1]x10-3) – a 15% striatal GSH deficit in PD (Figure 2). Also in the striatum, NAA/W in PD patients ([8.7±1.2]x10-2) was significant lower (p=0.04) than in the HV group ([9.4.x±1.0]x10-2) (Figure 3). In the OCC, a region not directly implicated in PD, there was a trend-level lower GSH/W (p=0.08) in PD patients ([1.8±0.4]x10-3) than in the HV group ([2.1±0.5]x10-3) (Figure 2). However, unlike in the striatum OCC NAA/W did not differ significantly (p=0.20) between patients with PD ([4.6±0.6]x10-2) and HV subjects ([4.5±0.9]x10-2) (Figure 3). None of the other major neurometabolites (tCr, tCho) differed between the groups either in the striatum or the OCC.
DISCUSSION and CONCLUSION
This study has obtained what is, perhaps, the very first direct experimental evidence of a significant nigrostriatal GSH deficit in vivo in PD compared to HV subjects (Figure 2), a finding that corroborates the results of studies in postmortem PD brain that have consistently reported abnormally low striatal GSH levels3-6 and have formed the basis for one of the current leading hypotheses implicating oxidative stress in PD pathophysiology. In addition, this study has found levels of the putative neuronal marker, NAA, to be lower in the striatum of PD patients than in HV (Figure 2). Along with our observation of normal OCC NAA levels in PD – despite a trend toward lower GSH in the same region that suggests that the effects of oxidative stress and associated consumption of antioxidant reserves are widespread – the finding of a selective striatal NAA deficit in PD (Figure 2) is consistent with both a nigrostriatal neurodegeneration and the established regional specificity of striatal abnormalities that are the hallmark of PD. In contrast, the OCC seems to remain relatively better preserved in PD. Lastly, the present finding of a significant striatal GSH deficit in PD patients in vivo provides a compelling rationale for investigating GSH synthetic precursors (e.g., N-acetylcysteine) that can be administered to spur in situ synthesis and elevation of cortical GSH for neuroprotection against oxidative stress.
Acknowledgements
No acknowledgement found.References
Seet RCS, Lee CYJ, Lim ECH, Tan JJH, Quek AML, Chong WL, Looi WF, Huang SH, Wang H, Chan UH, Halliwell B: Oxidative damage in Parkinson disease: Measurement using accurate biomarkers. Free Radical Biology and Medicine 2010, 48: 560-566.
Jenner P: Oxidative stress and Parkinson's disease. Handb Clin Neurol 2007, 83: 507-520.
Sofic E, Lange KW, Jellinger K, Riederer P: Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson's disease. Neurosci Lett 1992, 142(2): 128-130.
Jenner P, Dexter DT, Sian J, Schapira AH, Marsden CD: Oxidative stress as a cause of nigral cell death in Parkinson's disease and incidental Lewy body disease. The Royal Kings and Queens Parkinson's Disease Research Group. Ann Neurol 1992, 32 Suppl: S82-87.
Riederer P, Sofic E, Rausch WD, Schmidt B, Reynolds GP, Jellinger K, Youdim MB: Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J Neurochem 1989, 52(2): 515-520.
Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, Jenner P, Marsden CD: Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol 1994, 36(3): 348-355.
Zeevalk GD, Razmpour R, Bernard LP: Glutathione and Parkinson's disease: is this the elephant in the room? Biomed Pharmacother 2008, 62: 236-249.
Figures
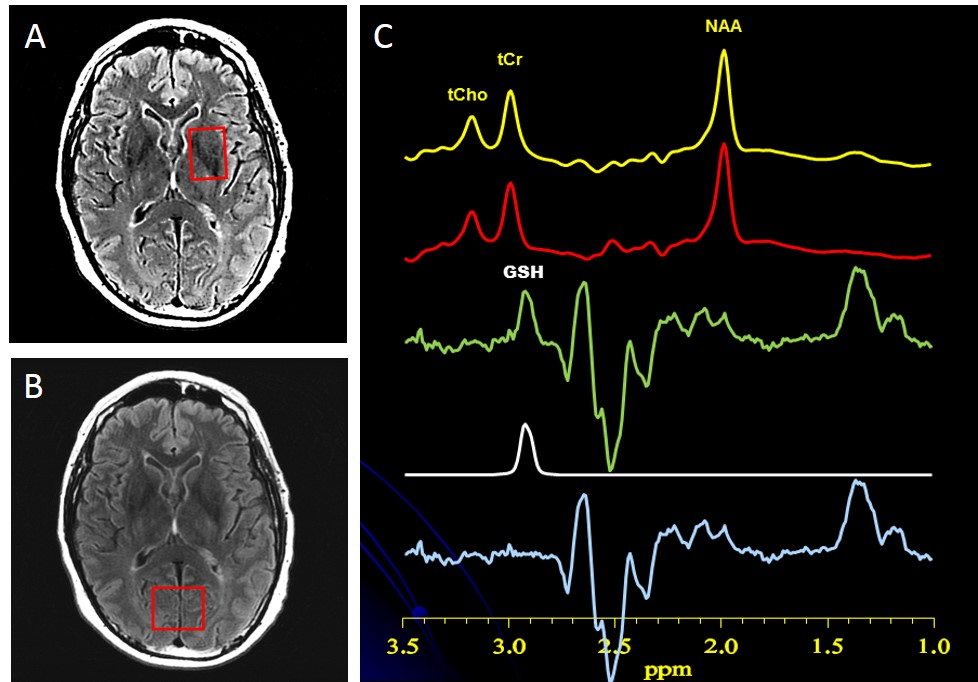
Figure 1: A: Location and size of striatal voxel. B: Location and size of OCC voxel. C: Illustration of brain GSH detection by J-edited difference 1H MRS, followed by frequency-domain fitting of the difference spectrum (c) to obtain GSH peak areas that yield GSH levels.
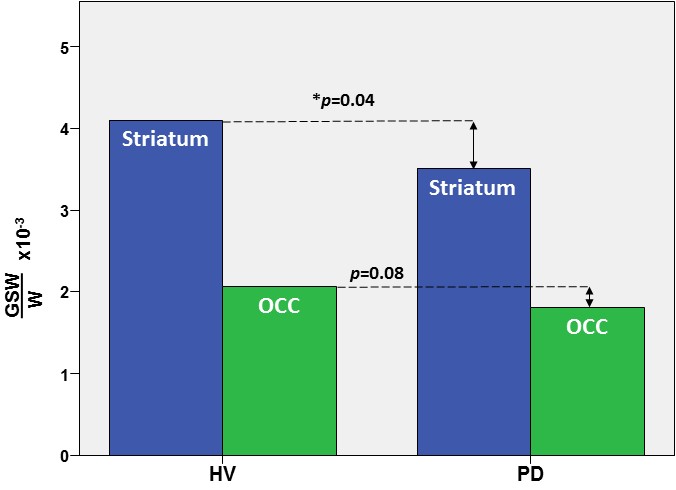
Figure 2: In vivo GSH levels in the striatum (blue) and occipital cortex (green) in patients with PD and in healthy volunteer (HV) subjects measured with J-edited 1H MRS.
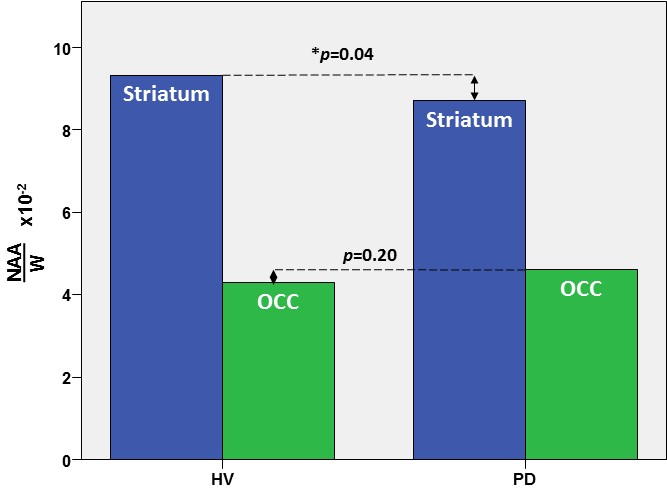
Figure 3: In vivo GSH levels in the striatum (blue) and occipital cortex (green) in patients with PD and in healthy volunteer (HV) subjects measured with J-edited 1H MRS.